技术领域
本发明属于有机化合物合成技术领域,具体涉及一种环氧查尔酮及其衍生物的合成方法。
背景技术
环氧化合物是有机合成的重要中间体和有机化工原料,如环氧丙烷、环氧氯丙烷、环氧苯乙烷等。它们在石油化工、高分子合成材料、精细化工、有机合成和制药等领域具有广泛应用。传统合成环氧化合物主要有以下几种方法:方法一是氯代醇法,将烯烃与次氯酸或次氯酸钙反应得到氯代醇,再与碱反应生成环氧化合物(CN103058142A;US4341756)。该法使用有毒的氯气,对设备腐蚀性大,存在安全隐患,而且其会产生大量的工业废料。方法二是有机过氧化物氧化法,将烯烃与有机过氧化物混合在一定条件下反应得到环氧化合物(US4284565A)。该法使用的有机过氧化合物不稳定,易燃易爆,存在安全隐患。方法三是金属催化剂/过氧化氢氧化法,将烯烃、金属催化剂和过氧化氢混合反应制得环氧化合物(CN102666516A)。该法使用有毒且昂贵的金属催化剂,而且过氧化氢具有强氧化性易发生爆炸。
因此,发展一种新型的氧化方法来制备环氧化合物具有很大的潜在价值。
发明内容
为解决目前制备环氧化合物中存在安全隐患的问题,本发明提出了一种环氧查尔酮及其衍生物的合成方法,本方法安全易行,而且具有催化剂价廉易得且毒性小,反应条件温和,官能团普适性好及操作简便等优点。
本发明是通过以下技术方案实现的:一种环氧查尔酮及其衍生物的合成方法,以取代查尔酮为起始物,在催化剂和氧化剂的存在下,在溶剂中于0~50℃下反应0.5~6小时,制备环氧查尔酮及其衍生物。
制备环氧查尔酮及其衍生物的反应式如下所示:
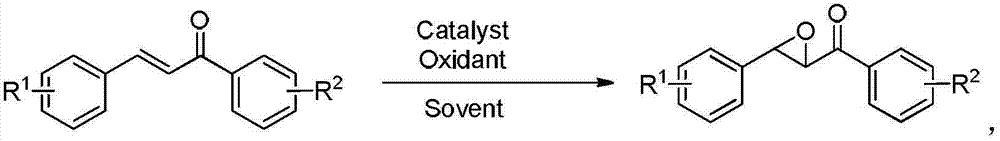
式中R1、R2分别独立选自H、甲基、乙基、甲氧基、乙氧基、氟、氯、溴、甲酰基、乙氧羰基、硝基中的一种或几种。
所述的催化剂选自铜粉、碘化亚铜、溴化亚铜、氯化亚铜、醋酸铜、硫酸铜中的一种或几种,催化剂的用量为取代查尔酮物质的量的1%~30%,优选量为10%。
所述氧化剂选自1-氯甲基-4-氟-1,4-二氮杂双环[2.2.2]辛烷二(四氟硼酸)盐)、N-氟-二苯磺酰胺、N-氟吡啶三氟甲磺酸盐中的一种或几种,氧化剂用量为取代查尔酮物质的量的100%~300%,优选量为200%。
所述溶剂为乙腈与水的混合溶液,溶剂的量为使溶质溶解的量。作为优选,混合溶液中乙腈与水的体积比为100~600∶1,更优选体积比为400∶1。
作为优选,本发明所述的反应中反应温度为室温,室温为25℃。反应时间为4小时。
本发明具体推荐所述的环氧查尔酮及其衍生物的合成方法包括如下步骤:将查尔酮、催化剂、氧化剂和乙腈/水(V∶V=400∶1)加入反应容器中,于0~50℃下搅拌反应0.5~6小时,所得反应液经分离纯化得到目标产物。
进一步,所述的分离纯化可采用如下方法:所得反应液中加入柱层析硅胶,并通过减压蒸馏除去溶剂,再通过TLC(以石油醚/乙酸乙酯体积比=20∶1作为洗脱剂)分离得到纯产物。
与现有技术相比,本发明的有益效果是:
(1)催化剂价廉易得,毒性较低;
(2)使用的氧化剂稳定,不存在易燃易爆等安全隐患;
(3)反应条件较温和,节约能源消耗;
(4)还具有产率高,底物普适性强,操作简便等特点。
具体实施方式
下面通过实施例对本发明作进一步详细说明,实施例中所述的室温为25℃。
实施例1
将查尔酮(0.2 mmol)、Selectfluor(0.4 mmol)和铜粉(0.02 mmol)加入到10 mL圆底烧瓶中,再加入2 mL乙腈/水(V∶V=400∶1)作溶剂。接着,于室温条件下磁力搅拌4 h。然后,在反应液中加入两药匙柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,再通过柱色谱分离得到产物纯品(以石油醚/乙酸乙酯体积比=20∶1作为洗脱剂)。该物质为黄色固体,产率92%。
表征数据:mp:89-91℃;1H NMR(500 MHz,CDCl3)δ8.03(dd,J=8.2,1.0 Hz,2H),7.64(t,J=7.4 Hz,1H),7.51(t,J=7.8 Hz,2H),7.44-7.38(m,5H),4.32(d,J=1.9Hz,1H),4.10(d,J=1.8 Hz,1H).13CNMR(125MHz,)δ193.09,135.51,133.97,129.05,128.83(d,J=12.7 Hz),128.36,125.80,61.04,59.36;GC-MS(ESI,70eV):m/z(%)=224(100)[M+].
实施例2
将查尔酮(0.2 mmol)、Selectfluor(0.6 mmol)和碘化亚铜(0.002 mmol)加入到10 mL圆底烧瓶中,再加入2 mL乙腈/水(V∶V=400∶1)作溶剂。接着,于0℃下磁力搅拌6 h。然后,在反应液中加入两药匙柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,再通过柱色谱分离得到产物纯品(以石油醚/乙酸乙酯体积比=20∶1作为洗脱剂)。该物质为黄色固体,产率85%。

表征数据:mp:89-91℃;1H NMR(500 MHz,CDCl3)δ8.03(dd,J=8.2,1.0 Hz,2H),7.64(t,J=7.4 Hz,1H),7.51(t,J=7.8 Hz,2H),7.44-7.38(m,5H),4.32(d,J=1.9Hz,1H),4.10(d,J=1.8 Hz,1H).13CNMR(125MHz,)δ193.09,135.51,133.97,129.05,128.83(d,J=12.7 Hz),128.36,125.80,61.04,59.36;GC-MS(ESI,70eV):m/z(%)=224(100)[M+].
实施例3
将查尔酮(0.2 mmol)、Selectfluor(0.2 mmol)和溴化亚铜(0.02 mmol)加入到10 mL圆底烧瓶中,再加入2 mL乙腈/水(V∶V=100∶1)作溶剂。接着,于50℃下磁力搅拌4 h。然后,在反应液中加入两药匙柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,再通过柱色谱分离得到产物纯品(以石油醚/乙酸乙酯体积比=20∶1作为洗脱剂)。该物质为黄色固体,产率88%。
表征数据:mp:89-91℃;1H NMR(500 MHz,CDCl3)δ8.03(dd,J=8.2,1.0 Hz,2H),7.64(t,J=7.4 Hz,1H),7.51(t,J=7.8 Hz,2H),7.44-7.38(m,5H),4.32(d,J=1.9Hz,1H),4.10(d,J=1.8 Hz,1H).13CNMR(125MHz,)δ193.09,135.51,133.97,129.05,128.83(d,J=12.7 Hz),128.36,125.80,61.04,59.36;GC-MS(ESI,70eV):m/z(%)=224(100)[M+].
实施例4
将查尔酮(0.2 mmol)、N-氟吡啶三氟甲磺酸盐(0.4 mmol)和醋酸铜(0.06 mmol)加入到10 mL圆底烧瓶中,再加入2 mL乙腈/水(V∶V=600∶1)作溶剂。接着,于室温条件下磁力搅拌3 h。然后,在反应液中加入两药匙柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,再通过柱色谱分离得到产物纯品(以石油醚/乙酸乙酯体积比=20∶1作为洗脱剂)。该物质为黄色固体,产率90%。
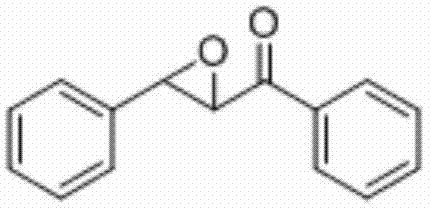
表征数据:mp:89-91℃;1H NMR(500 MHz,CDCl3)δ8.03(dd,J=8.2,1.0 Hz,2H),7.64(t,J=7.4 Hz,1H),7.51(t,J=7.8 Hz,2H),7.44-7.38(m,5H),4.32(d,J=1.9Hz,1H),4.10(d,J=1.8 Hz,1H).13CNMR(125MHz,)δ193.09,135.51,133.97,129.05,128.83(d,J=12.7 Hz),128.36,125.80,61.04,59.36;GC-MS(ESI,70eV):m/z(%)=224(100)[M+].
实施例5
将查尔酮(0.2 mmol)、N-氟-二苯磺酰胺(0.4 mmo)和硫酸铜(0.004 mmol)加入到10 mL圆底烧瓶中,再加入2 mL乙腈/水(V∶V=500∶1)作溶剂。接着,于45℃下磁力搅拌0.5 h。然后,在反应液中加入两药匙柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,再通过柱色谱分离得到产物纯品(以石油醚/乙酸乙酯体积比=20∶1作为洗脱剂)。该物质为黄色固体,产率82%。
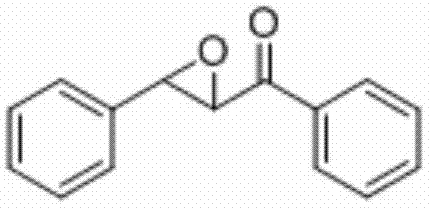
表征数据:mp:89-91℃;1H NMR(500 MHz,CDCl3)δ8.03(dd,J=8.2,1.0 Hz,2H),7.64(t,J=7.4 Hz,1H),7.51(t,J=7.8 Hz,2H),7.44-7.38(m,5H),4.32(d,J=1.9 Hz,1H),4.10(d,J=1.8 Hz,1H).13CNMR(125MHz,)δ193.09,135.51,133.97,129.05,128.83(d,J=12.7 Hz),128.36,125.80,61.04,59.36;GC-MS(ESI,70eV):m/z(%)=224(100)[M+].
实施例6
将查尔酮(0.2 mmol)、Selectfluor(0.5 mmol)和铜粉(0.03 mmol)加入到10 mL圆底烧瓶中,再加入2 mL乙腈/水(V∶V=200∶1)作溶剂。接着,于室温条件下磁力搅拌2 h。然后,在反应液中加入两药匙柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,再通过柱色谱分离得到产物纯品(以石油醚/乙酸乙酯体积比=20∶1作为洗脱剂)。该物质为黄色固体,产率90%。
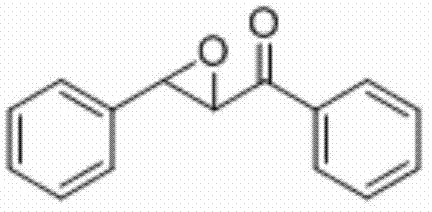
表征数据:mp:89-91℃;1H NMR(500 MHz,CDCl3)δ8.03(dd,J=8.2,1.0 Hz,2H),7.64(t,J=7.4 Hz,1H),7.51(t,J=7.8 Hz,2H),7.44-7.38(m,5H),4.32(d,J=1.9Hz,1H),4.10(d,J=1.8 Hz,1H).13CNMR(125MHz,)δ193.09,135.51,133.97,129.05,128.83(d,J=12.7 Hz),128.36,125.80,61.04,59.36;GC-MS(ESI,70eV):m/z(%)=224(100)[M+].
实施例7
将3-(4-氯苯基)-1-苯基丙-2-烯-1-酮(0.2 mmol)、Selectfluor(0.4 mmol)和铜粉(0.02 mmol)加入到10 mL圆底烧瓶中,再加入2 mL乙腈/水(V∶V=400∶1)作溶剂。接着,于室温条件下磁力搅拌3 h。然后,在反应液中加入两药匙柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,再通过柱色谱分离得到产物纯品(以石油醚/乙酸乙酯体积比=20∶1作为洗脱剂)。该物质为黄色固体,产率82%。
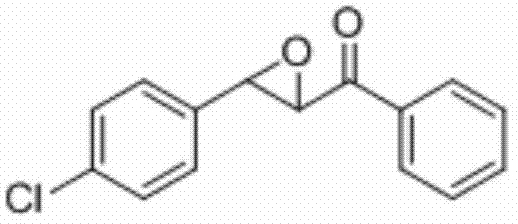
表征数据:mp:70-73℃;1H NMR(500 MHz,CDCl3)δ8.04-8.00(m,2H),7.67-7.63(m,1H),7.52(dd,J=10.8,4.8 Hz,2H),7.42-7.38(m,2H),7.35-7.31(m,2H),4.27(d,J=1.9 Hz,1H),4.08(d,J=1.8 Hz,1H).GC-MS(ESI,70eV):m/z(%)=258(100)[M+].
实施例8
将3-(4-氯苯基)-1-苯基丙-2-烯-1-酮(0.2 mmol)、Selectfluor(0.3 mmo)和碘化亚铜(0.05 mmol)加入到10 mL圆底烧瓶中,再加入2 mL乙腈/水(V∶V=300∶1)作溶剂。接着,于35℃下磁力搅拌5 h。然后,在反应液中加入两药匙柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,再通过柱色谱分离得到产物纯品(以石油醚/乙酸乙酯体积比=20∶1作为洗脱剂)。该物质为黄色固体,产率80%。
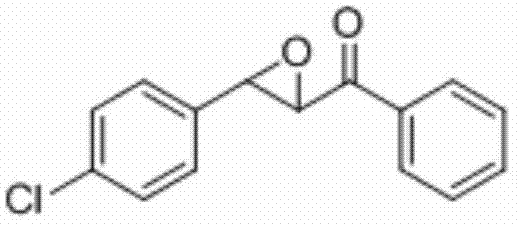
表征数据:mp:70-73℃;1H NMR(500 MHz,CDCl3)δ8.04-8.00(m,2H),7.67-7.63(m,1H),7.52(dd,J=10.8,4.8 Hz,2H),7.42-7.38(m,2H),7.35-7.31(m,2H),4.27(d,J=1.9 Hz,1H),4.08(d,J=1.8 Hz,1H).GC-MS(ESI,70eV):m/z(%)=258(100)[M+].
实施例9
将3-(4-氯苯基)-1-苯基丙-2-烯-1-酮(0.2 mmol)、N-氟-二苯磺酰胺(0.5 mmo)和硫酸铜(0.01 mmol)加入到10 mL圆底烧瓶中,再加入2 mL乙腈/水(V∶V=100∶1)作溶剂。接着,于45℃下磁力搅拌4 h。然后,在反应液中加入两药匙柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,再通过柱色谱分离得到产物纯品(以石油醚/乙酸乙酯体积比=20∶1作为洗脱剂)。该物质为黄色固体,产率78%。
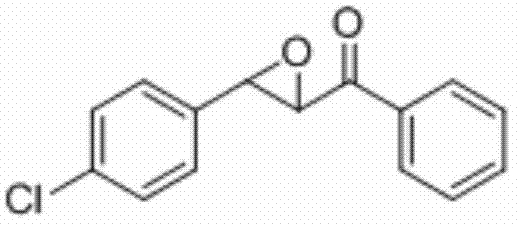
表征数据:mp:70-73℃;1H NMR(500 MHz,CDCl3)δ8.04-8.00(m,2H),7.67-7.63(m,1H),7.52(dd,J=10.8,4.8 Hz,2H),7.42-7.38(m,2H),7.35-7.31(m,2H),4.27(d,J=1.9 Hz,1H),4.08(d,J=1.8 Hz,1H).GC-MS(ESI,70eV):m/z(%)=258(100)[M+].
实施例10
将1-(4-溴苯基)-3-(4-氯苯基)丙-2-烯-1-酮(0.2 mmol)、Selectfluor(0.4 mmol)和铜粉(0.02 mmol)加入到10 mL圆底烧瓶中,再加入2 mL乙腈/水(V∶V=400∶1)作溶剂。接着,于室温条件下磁力搅拌4 h。然后,在反应液中加入两药匙柱层析硅 胶(100-200目),并通过减压蒸馏除去溶剂,再通过柱色谱分离得到产物纯品(以石油醚/乙酸乙酯体积比=20∶1作为洗脱剂)。该物质为黄色固体,产率90%。
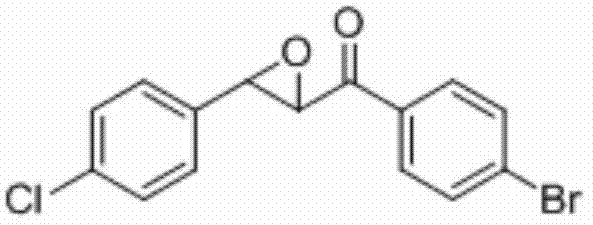
表征数据:mp:108-111℃;1H NMR(500 MHz,)δ7.89(d,J=8.6 Hz,2H),7.66(d,J=8.5 Hz,2H),7.40(d,J=8.5 Hz,2H),7.31(d,J=8.5 Hz,2H),4.19(d,J=1.8 Hz,1H),4.07(d,J=1.7 Hz,1H).;13C NMR(125MHz,CDCl3)δ191.96,135.12,134.01,133.81,132.32,129.87,129.55,129.10,127.12,60.99,58.68;GC-MS(ESI,70eV):m/z(%)=235(100)[M+].
实施例11
将1-(4-溴苯基)-3-(4-氯苯基)丙-2-烯-1-酮(0.2 mmol)、N-氟吡啶三氟甲磺酸盐(0.5 mmol)和醋酸铜(0.04 mmol)加入到10 mL圆底烧瓶中,再加入2 mL乙腈/水(V∶V=100∶1)作溶剂。接着,于室温条件下磁力搅拌4 h。然后,在反应液中加入两药匙柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,再通过柱色谱分离得到产物纯品(以石油醚/乙酸乙酯体积比=20∶1作为洗脱剂)。该物质为黄色固体,产率88%。
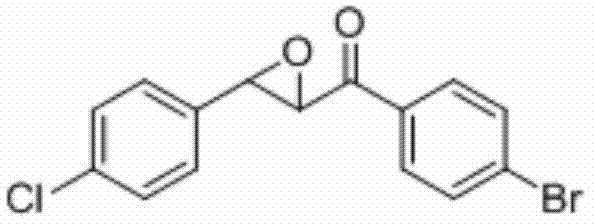
表征数据:mp:108-111℃;1H NMR(500 MHz,)δ7.89(d,J=8.6 Hz,2H),7.66(d,J=8.5 Hz,2H),7.40(d,J=8.5 Hz,2H),7.31(d,J=8.5 Hz,2H),4.19(d,J=1.8 Hz,1H),4.07(d,J=1.7 Hz,1H).;13C NMR(125MHz,CDCl3)δ191.96,135.12,134.01,133.81,132.32,129.87,129.55,129.10,127.12,60.99,58.68;GC-MS(ESI,70eV):m/z(%)=235(100)[M+].
实施例12
将1-(4-溴苯基)-3-(4-氯苯基)丙-2-烯-1-酮(0.2 mmol)、Selectfluor(0.2 mmol)和氯化亚铜(0.06 mmol)加入到10 mL圆底烧瓶中,再加入2 mL乙腈/水(V∶V=300∶1)作溶剂。接着,于室温条件下磁力搅拌4 h。然后,在反应液中加入两药匙柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,再通过柱色谱分离得到产物纯 品(以石油醚/乙酸乙酯体积比=20∶1作为洗脱剂)。该物质为黄色固体,产率85%。
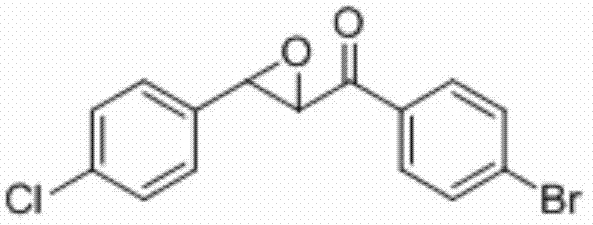
表征数据:mp:108-111℃;1H NMR(500 MHz,)δ7.89(d,J=8.6 Hz,2H),7.66(d,J=8.5 Hz,2H),7.40(d,J=8.5 Hz,2H),7.31(d,J=8.5 Hz,2H),4.19(d,J=1.8 Hz,1H),4.07(d,J=1.7 Hz,1H).;13C NMR(125MHz,CDCl3)δ191.96,135.12,134.01,133.81,132.32,129.87,129.55,129.10,127.12,60.99,58.68;GC-MS(ESI,70eV):m/z(%)=235(100)[M+].
实施例13
将3-苯基-1-(对甲苯基)丙-2-烯-1-酮(0.2 mmol)、Selectfluor(0.4 mmo)和铜粉(0.02mmol)加入到10 mL圆底烧瓶中,再加入2 mL乙腈/水(V∶V=400∶1)作溶剂。接着,于室温条件下磁力搅拌4 h。然后,在反应液中加入两药匙柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,再通过柱色谱分离得到产物纯品(以石油醚/乙酸乙酯体积比=20∶1作为洗脱剂)。该物质为黄色固体,产率80%。
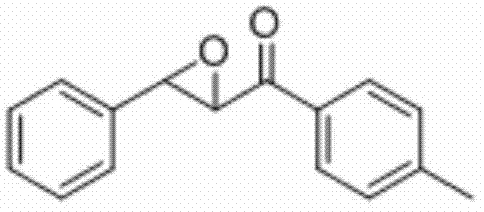
表征数据:mp:85-89℃;1H NMR(500 MHz,)δ7.93(d,J=8.2 Hz,2H),7.46-7.37(m,5H),7.30(d,J=8.1 Hz,2H),4.30(d,J=1.8 Hz,1H),4.09(d,J=1.7 Hz,1H),2.44(s,3H).13C NMR(125MHz,CDCl3)δ192.61,145.06,135.64,133.07,129.56,128.99,128.75,128.48,125.80,60.94,59.28,21.76;GC-MS(ESI,70eV):m/z(%)=238(100)[M+].
实施例14
将3-苯基-1-(对甲苯基)丙-2-烯-1-酮(0.2 mmol)、N-氟-二苯磺酰胺(0.5 mmo)和铜粉(0.04 mmol)加入到10 mL圆底烧瓶中,再加入2 mL乙腈/水(V∶V=200∶1)作溶剂。接着,于室温条件下磁力搅拌4 h。然后,在反应液中加入两药匙柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,再通过柱色谱分离得到产物纯品(以石油醚/乙酸乙酯体积比=20∶1作为洗脱剂)。该物质为黄色固体,产率79%。
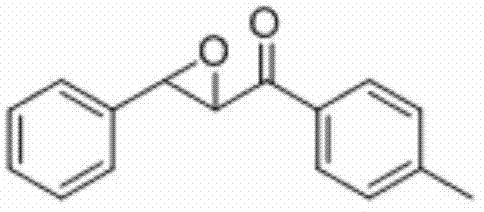
表征数据:mp:85-89℃;1H NMR(500 MHz,)δ7.93(d,J=8.2 Hz,2H),7.46-7.37(m,5H),7.30(d,J=8.1 Hz,2H),4.30(d,J=1.8 Hz,1H),4.09(d,J=1.7 Hz,1H),2.44(s,3H).13C NMR(125MHz,CDCl3)δ192.61,145.06,135.64,133.07,129.56,128.99,128.75,128.48,125.80,60.94,59.28,21.76;GC-MS(ESI,70eV):m/z(%)=238(100)[M+].
实施例15
将1,3-二(4-氯苯基)丙-2-烯-1-酮(0.2 mmol)、Selectfluor(0.5 mmo)和碘化亚铜(0.03 mmol)加入到10 mL圆底烧瓶中,再加入2 mL乙腈/水(V∶V=200∶1)作溶剂。接着,于室温条件下磁力搅拌5 h。然后,在反应液中加入两药匙柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,再通过柱色谱分离得到产物纯品(以石油醚/乙酸乙酯体积比=20∶1作为洗脱剂)。该物质为黄色固体,产率89%。
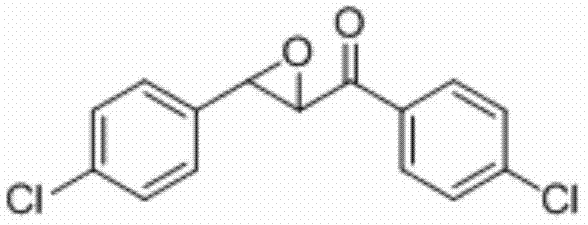
表征数据:mp:98-101℃;1H NMR(500 MHz,CDCl3)δ7.99-7.96(m,2H),7.50-7.47(m,2H),7.41-7.38(m,2H),7.32-7.30(m,2H),4.20(d,J=1.8 Hz,1H),4.07(d,J=1.7 Hz,1H);13CNMR(125MHz)δ191.74,140.77,135.12,133.85,133.64,129.82,129.32,129.10,127.12,61.02,58.66;GC-MS(ESI,70eV):m/z(%)=280(100)[M+].
实施例16
将1,3-二(4-氯苯基)丙-2-烯-1-酮(0.2 mmol)、N-氟吡啶三氟甲磺酸盐(0.5 mmol)和铜粉(0.02 mmol)加入到10 mL圆底烧瓶中,再加入2 mL乙腈/水(V∶V=300∶1)作溶剂。接着,于室温条件下磁力搅拌4 h。然后,在反应液中加入两药匙柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,再通过柱色谱分离得到产物纯品(以石油醚/乙酸乙酯体积比=20∶1作为洗脱剂)。该物质为黄色固体,产率85%。
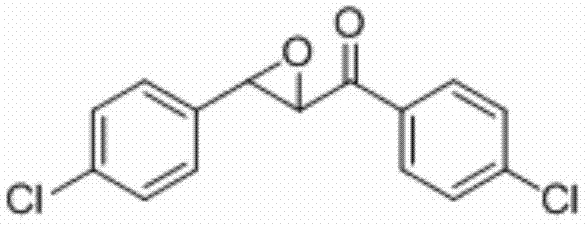
表征数据:mp:98-101℃;1H NMR(500 MHz,CDCl3)δ7.99-7.96(m,2H),7.50-7.47(m,2H),7.41-7.38(m,2H),7.32-7.30(m,2H),4.20(d,J=1.8 Hz,1H),4.07(d,J=1.7 Hz,1H);13CNMR(,125MHz)δ191.74,140.77,135.12,133.85,133.64,129.82,129.32,129.10,127.12,61.02,58.66;GC-MS(ESI,70eV):m/z(%)=280(100)[M+].
实施例17
将1-苯基-3-(对甲苯基)丙-2-烯-1-酮(0.2 mmol)、Selectfluor(0.4 mmo)和铜粉(0.02 mmol)加入到10 mL圆底烧瓶中,再加入2 mL乙腈/水(V∶V=400∶1)作溶剂。接着,于室温条件下磁力搅拌4 h。然后,在反应液中加入两药匙柱层析硅胶(100-200目),并通过减压蒸馏除去溶剂,再通过柱色谱分离得到产物纯品(以石油醚/乙酸乙酯体积比=20∶1作为洗脱剂)。该物质为黄色固体,产率64%。

表征数据:mp:70-76℃;1H NMR(500 MHz,CDCl3)δ8.03-8.01(m,2H),7.65-7.61(m,1H),7.55-7.45(m,3H),7.29(s,1H),7.23(d,J=8.1 Hz,2H),4.31(d,J=2.0 Hz,1H),4.06(d,J=2.0 Hz,1H),2.40(s,3H).13C NMR(125MHz,CDCl3)δ193.22,139.07,135.53,133.94,132.47,129.47,128.86,128.34,125.78,61.08,59.45,21.26;GC-MS(ESI,70eV):m/z(%)=238(100)[M+]。

 手机二维码
手机二维码 京公网安备11011302007134
京公网安备11011302007134