技术领域
本发明涉及催化化学领域,具体涉及一种用于合成手性二级醇的催化剂,其制备方法和采用该催化剂的手性二级醇合成方法。
背景技术
手性二级醇尤其手性芳香二级醇是高科技、高附加值产品,它们可分别作为合成(R)-氟西汀、(R)-沙丁胺醇等多种治疗神经系统、心血管等疾病的手性药物的重要中间体。因此,手性二级醇的研发,对推进和实践科学发展有着重要的作用。
目前,手性二级醇通常经由潜手性酮不对称加氢、氢转移氢化、手性硼杂噁唑烷催化还原等方法来制备。潜手性酮不对称硅氢化是不对称加氢的重要方法之一,与使用氢气相比,使用氢硅烷作为氢源,其主要的优点是条件特别的温和,无需耐高压技术,可操作性强。潜手性酮的不对称硅氢加成反应,可以简单、高效地制备得到手性二级醇。经过多年研究和探索,人们已经研发出了多种用于潜手性酮不对称硅氢加成反应的金属催化剂,1973年Kagan等(Dumont,W.;Poulin,J.C.;Phat,T.D.;Kagan,H.B.J.Am.Chem.Soc.1973,95,8295)首次报道Rh(I)/DIOP催化潜手性酮和亚胺不对称硅氢加成。此后,大量的研究集中在高效催化剂的制备上。早期的不对称硅氢加成反应催化剂主要局限于使用Rh,Ir和Ru等贵金属,存在着催化剂成本较高,难以重复利用,以及催化剂流失等问题。后来发现过渡金属催化剂如Zn,Cu,Fe等新型催化剂也能催化不对称硅氢加成反应,并且发现这些过渡金属催化剂在特定条件下对酮的硅氢加成反应也具有高活性和立体选择性。如中国专利CN201210454160.4报道以环己二胺出发合成手性配体,然后络合锌得到催化剂用于潜手性酮的不对称硅氢加成反应。但是环己二胺价格较贵,存在拆分等繁琐步骤。因此,研发出新颖的、绿色环保、廉价的、可操作性强的、具有高立体选择性手性配体仍然是今后研究的重要方向。
发明内容
为了解决目前的用于手性二级醇的催化剂存在的成本较高、重复利用率低拆分繁琐等问题,提供一种用于合成手性二级醇的催化剂,其制备方法和采用该催化剂的手性二级醇合成方法。
解决上述问题的具体方案为:
一种用于合成手性二级醇的催化剂,所述手性配体的结构式为:
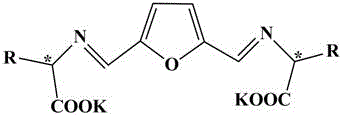 ,
,
结构式中:
R为甲基、异丙基、异丁基、仲丁基或苄基。
作为优选,所述R为甲基或苄基。
作为优选,所述催化剂为二乙基锌和手性配体反应生成的络合物,所述手性配体为采用呋喃二醛、氢氧化钾和氨基酸为原料化学反应生成的希夫碱。
一种用于合成手性二级醇的催化剂的制备方法,具体包括以下步骤:
1)容器中,以物质的量计,加入1份5-羟甲基糠醛与3-4份二氧化锰,并以二氯甲烷为反应溶剂,搅拌下回流8-20小时,过滤,滤液除去溶剂,产物采用水重结晶得中间体2,5-呋喃二甲醛;
2)容器中,以物质的量计,加入1份氨基酸、1份KOH,以乙醇为反应溶剂,10-50℃下搅拌30min-2h,然后再加入0.5份步骤1)制备的2,5-呋喃二甲醛,10-50℃下继续搅拌1-30小时后,停止反应,去除溶剂得到手性配体;
3)容器中,以物质的量计,加入1份步骤2)得到的手性配体,以正己烷为反应溶剂,然后加入1-2份二乙基锌,常温下搅拌2-12小时,除去溶剂得用于合成手性二级醇的催化剂。
作为优选,步骤2)所述氨基酸为D-缬氨酸、L-缬氨酸、D-丙氨酸、L-丙氨酸、D-亮氨酸、L-亮氨酸、D-异亮氨酸、L-异亮氨酸、D-苯丙氨酸或L-苯丙氨酸。
一种采用上述催化剂的手性二级醇合成方法,包括以下步骤:以物质的量计,容器中,依次加入1份催化剂、10-100份潜手性酮和溶剂,搅拌5-30分钟,然后加入10-300份含氢硅烷,25-60℃反应8-72小时,然后加入酸溶液或者碱溶液,水解2-20小时,分出有机相,除去溶剂,得手性二级醇。
作为优选,所述溶剂为四氢呋喃、甲苯或二氯甲烷。
作为优选,所述潜手性酮为苯乙酮、苯丙酮、苯丁酮、邻氯苯乙酮、对氯苯乙酮、对甲基苯乙酮、间三氟甲基苯乙酮、邻溴苯乙酮、四氢奈酮、6-甲氧基-1-茚酮或6-氯-1-茚酮。
作为优选,所述含氢硅烷为三甲氧基氢硅烷、三甲基氢硅烷、三乙氧基氢硅烷、三乙基氢硅烷或三苯基氢硅烷。
作为优选,所述酸溶液中H+或者碱溶液中OH-与潜手性酮等摩尔当量。
本发明与现有技术相比,有益效果是:
1本发明的催化剂用于潜手性酮硅氢加成时反应条件温和、安全、有效;
2本发明的催化剂是从天然产物的衍生物5-羟甲基糠醛出发而合成的,具有催化活性高、反应条件温和、成本低等特点。既有效开发了生物质的下游产品,又为不对称硅氢加成的催化剂增加了一个新品种;
3本发明的催化剂可以进一步水解得到手性二级醇;
4本发明的催化剂可控性强,重现性好,有明显的经济社会效益;
5本发明的催化剂的制备方法简单实用,为产品开发和催化剂设计提供了新思路。
具体实施方式
下面通过具体实施例对本发明的技术方案作进一步描述说明。
如果无特殊说明,本发明的实施例中所采用的原料均为本领域常用的原料,实施例中所采用的方法,均为本领域的常规方法。
实施例1
催化剂制备:容器中,加入1.26g5-羟甲基糠醛与3.4g二氧化锰,二氯甲烷为反应溶剂,搅拌下回流10小时,过滤,滤液除去溶剂,产物用水重结晶得中间体呋喃二甲醛。
容器中,加入0.585gD-缬氨酸、0.29gKOH以及20mL乙醇,25℃下搅拌至溶液透明。然后加入0.31g呋喃二甲醛,25℃下继续搅拌4小时。停止反应,去除溶剂得手性配体。
容器中加入0.398g手性配体,10mL正己烷,然后加入0.15mL二乙基锌(1mol/L,溶剂为正己烷),常温下搅拌2小时以上得催化剂。
实施例2:
催化剂制备:反应过程同实施例1,氨基酸换为L-缬氨酸。
实施例3:
催化剂制备:反应过程同实施例1,氨基酸换为D-丙氨酸。
实施例4:
催化剂制备:反应过程同实施例1,氨基酸换为L-丙氨酸。
实施例5:
催化剂制备:反应过程同实施例1,氨基酸换为D-亮氨酸。
实施例6:
催化剂制备:反应过程同实施例1,氨基酸换为L-亮氨酸。
实施例7:
催化剂制备:反应过程同实施例1,氨基酸换为D-异亮氨酸。
实施例8:
催化剂制备:反应过程同实施例1,氨基酸换为L-异亮氨酸。
实施例9:
催化剂制备:反应过程同实施例1,氨基酸换为D-苯丙氨酸。
实施例10:
催化剂制备:反应过程同实施例1,氨基酸换为L-苯丙氨酸。
实施例11:
手性二级醇的合成:容器中,加入0.0025g由实施例1所得催化剂,加入四氢呋喃6mL,加入0.1g苯乙酮,搅拌15分钟,然后加入0.3mL三乙氧基氢硅烷,25℃反应24小时,催化活性99%。加入与苯乙酮等摩尔量的KOH水溶液(质量百分比15%),水解3小时,分出有机相,干燥。有机相通过减压蒸馏手机馏分,得到1-苯基乙醇,光学选择性59%ee。
实施例12:
手性二级醇的合成:容器中,加入0.002g由实施例2所得催化剂,加入四氢呋喃6mL,加入0.1g苯乙酮,搅拌15分钟,然后加入0.3mL三乙氧基氢硅烷,25℃反应24小时,催化活性98%。加入与苯乙酮等摩尔量的KOH水溶液(质量百分比15%),水解3小时,分出有机相,干燥。有机相通过减压蒸馏手机馏分,得到1-苯基乙醇,光学选择性61%ee。
实施例13:
手性二级醇的合成:容器中,加入0.0021g由实施例3所得催化剂,加入四氢呋喃6mL,加入0.1g苯乙酮,搅拌15分钟,然后加入0.3mL三乙氧基氢硅烷,25℃反应24小时,催化活性95%。加入与苯乙酮等摩尔量的KOH水溶液(质量百分比15%),水解3小时,分出有机相,干燥。有机相通过减压蒸馏手机馏分,得到1-苯基乙醇,光学选择性46%ee。
实施例14:
手性二级醇的合成:容器中,加入0.0021g由实施例4所得催化剂,加入四氢呋喃6mL,加入0.1g苯乙酮,搅拌15分钟,然后加入0.3mL三乙氧基氢硅烷,25℃反应24小时,催化活性93%。加入与苯乙酮等摩尔量的KOH水溶液(质量百分比15%),水解3小时,分出有机相,干燥。有机相通过减压蒸馏手机馏分,得到1-苯基乙醇,光学选择性39%ee。
实施例15:
手性二级醇的合成:容器中,加入0.0023g由实施例5所得催化剂,加入四氢呋喃6mL,加入0.1g苯乙酮,搅拌15分钟,然后加入0.3mL三乙氧基氢硅烷,25℃反应24小时,催化活性96%。加入与苯乙酮等摩尔量的KOH水溶液(质量百分比15%),水解3小时,分出有机相,干燥。有机相通过减压蒸馏手机馏分,得到1-苯基乙醇,光学选择性48%ee。
实施例16:
手性二级醇的合成:容器中,加入0.0023g由实施例6所得催化剂,加入四氢呋喃6mL,加入0.1g苯乙酮,搅拌15分钟,然后加入0.3mL三乙氧基氢硅烷,25℃反应24小时,催化活性91%。加入与苯乙酮等摩尔量的KOH水溶液(质量百分比15%),水解3小时,分出有机相,干燥。有机相通过减压蒸馏手机馏分,得到1-苯基乙醇,光学选择性37%ee。
实施例17:
手性二级醇的合成:容器中,加入0.0023g由实施例7所得催化剂,加入四氢呋喃6mL,加入0.1g苯乙酮,搅拌15分钟,然后加入0.3mL三乙氧基氢硅烷,25℃反应24小时,催化活性98%。加入与苯乙酮等摩尔量的KOH水溶液(质量百分比15%),水解3小时,分出有机相,干燥。有机相通过减压蒸馏手机馏分,得到1-苯基乙醇,光学选择性55%ee。
实施例18:
手性二级醇的合成:容器中,加入0.0023g由实施例8所得催化剂,加入四氢呋喃6mL,加入0.1g苯乙酮,搅拌15分钟,然后加入0.3mL三乙氧基氢硅烷,25℃反应24小时,催化活性95%。加入与苯乙酮等摩尔量的KOH水溶液(质量百分比15%),水解3小时,分出有机相,干燥。有机相通过减压蒸馏手机馏分,得到1-苯基乙醇,光学选择性58%ee。
实施例19:
手性二级醇的合成:容器中,加入0.0031g由实施例9所得催化剂,加入四氢呋喃6mL,加入0.1g苯乙酮,搅拌15分钟,然后加入0.3mL三乙氧基氢硅烷,25℃反应24小时,催化活性87%。加入与苯乙酮等摩尔量的KOH水溶液(质量百分比15%),水解3小时,分出有机相,干燥。有机相通过减压蒸馏手机馏分,得到1-苯基乙醇,光学选择性52%ee。
实施例20:
手性二级醇的合成:容器中,加入0.0031g由实施例10所得催化剂,加入四氢呋喃6mL,加入0.1g苯乙酮,搅拌15分钟,然后加入0.3mL三乙氧基氢硅烷,25℃反应24小时,催化活性83%。加入与苯乙酮等摩尔量的KOH水溶液(质量百分比15%),水解3小时,分出有机相,干燥。有机相通过减压蒸馏手机馏分,得到1-苯基乙醇,光学选择性62%ee。
实施例21:
手性二级醇的合成:容器中,加入0.0025g由实施例1所得催化剂,加入四氢呋喃6mL,加入0.11g苯丙酮,搅拌15分钟,然后加入0.3mL三乙氧基氢硅烷,25℃反应36小时,催化活性78%。加入与苯丙酮等摩尔量的KOH水溶液(质量百分比15%),水解3小时,分出有机相,干燥。有机相通过减压蒸馏手机馏分,得到1-苯基丙醇,光学选择性58%ee。
实施例22:
手性二级醇的合成:容器中,加入0.0025g由实施例1所得催化剂,加入四氢呋喃6mL,加入0.12g邻氯苯乙酮,搅拌15分钟,然后加入0.3mL三乙基氢硅烷,25℃反应40小时,催化活性81%。加入与邻氯苯乙酮等摩尔量的KOH水溶液(质量百分比15%),水解3小时,分出有机相,干燥。有机相通过减压蒸馏手机馏分,得到邻氯苯乙醇,光学选择性56%ee。
实施例23:
手性二级醇的合成:容器中,加入0.0025g由实施例1所得催化剂,加入四氢呋喃6mL,加入0.13g对甲氧基苯乙酮,搅拌15分钟,然后加入0.3mL三乙基氢硅烷,25℃反应40小时,催化活性73%。加入与甲氧基苯乙酮等摩尔量的KOH水溶液(质量百分比15%),水解3小时,分出有机相,干燥。有机相通过减压蒸馏手机馏分,得到1-对甲氧基苯乙醇,光学选择性50%ee。
实施例24:
手性二级醇的合成:容器中,加入0.0025g由实施例1所得催化剂,加入四氢呋喃6mL,加入0.16g邻溴苯乙酮,搅拌15分钟,然后加入0.3mL三乙基氢硅烷,25℃反应40小时,催化活性63%。加入与邻溴苯乙酮等摩尔量的KOH水溶液(质量百分比15%),水解3小时,分出有机相,干燥。有机相通过减压蒸馏手机馏分,得到1-对溴苯基乙醇,光学选择性69%ee。

 手机二维码
手机二维码 京公网安备11011302007134
京公网安备11011302007134