技术领域
本发明涉及一类药物的制备方法,更具体的说是香豆素类抗凝药 ——手性华法林及其衍生物的制备方法。
背景技术
华法林的化学名为苄丙酮香豆素,其结构如下I式:
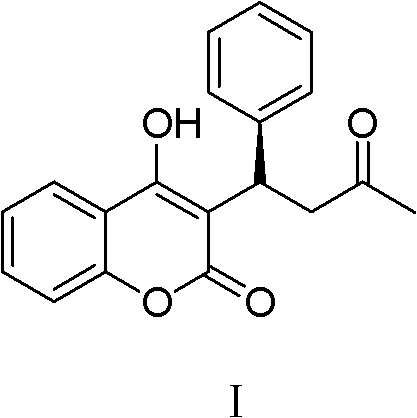
华法林为香豆素类口服抗凝药,临床用于预防和治疗血栓栓塞性 疾病,可防止血栓形成与发展,如治疗血栓栓塞性静脉炎,降低肺栓 塞的发病率和死亡率,减少外科大手术,风湿性心脏病、髋关节固定 术、人工置换心脏瓣膜手术等的静脉血栓发病率。此外,还可用于治 疗手术后或创伤后的静脉血栓形成,并可做心肌梗塞的辅助用药。多 种外科、介入手术后也须常规服用。
华法林主要经肝脏细胞色素P450(CYP)酶系代谢,能抑制CYP 活性的药物均可使华法林的代谢减慢,半衰期延长,抗凝作用增强; 反之亦然。华法林含2种异构体,S型经肝脏CYP2C9酶代谢,R 型由CYP3A4等酶代谢。血小板在止血、血栓形成、动脉粥样硬化 等过程中起着重要作用。药物主要通过抑制花生四烯酸代谢,增加血 小板内cAMP浓度等机制而抑制血小板粘附、聚集和分泌功能。
本药通过抑制维生素K在肝脏细胞内合成凝血因子II、VII、 IX、X,从而发挥抗凝作用。肝脏微粒体内的羧基化酶能将上述 凝血因子的谷氨酸转变为γ-羧基谷氨酸,后者再与钙离子结合, 才能发挥其凝血活性。本药的作用是抑制羧基化酶,对已经合成 的上述因子并无直接对抗作用,必须等待这些因子在体内相对耗 竭后,才能发挥抗凝效应,所以本药起效缓慢,仅在体内有效, 停药后药效持续时间较长(直到维生素K依赖性因子逐渐恢复 到一定浓度后,抗凝作用才消失)。此外,本药尚能诱导肝脏产生 维生素K依赖性凝血因子前体物质,并使之释放入血,该物质 抗原性与有关凝血因子相同,但并无凝血功能,反而具有抗凝血 作用,并能降低凝血酶诱导的血小板聚集反应。因此,在本药作 用下,凝血因子II、VII、IX、X、蛋白S和蛋白C合成减少, 而″假凝血因″亦即″维生素K拮抗药诱导蛋白质″增多,达到抗凝 效应。本药的药动学参数较稳定,优于其它口服抗凝药(如茴茚二 酮、苯丙羟香豆素和双香豆素等)。只有当患者对本药不耐受时, 才选用其它口服抗凝药。在非风湿性心房颤动患者预防脑卒中时, 本药疗效明显优于阿司匹林。在治疗或预防妊娠患者血栓或栓塞 形成时,皮下或静脉注射肝素疗效则优于本药(因肝素易通过胎 盘)。
本药是香豆素类抗凝剂的一种,在体内有对抗维生素K的作 用。可以抑制维生素K参与的凝血因子II、VII、IX、X的在肝脏 的合成。对血液中已有的凝血因子II、VII、IX、X并无抵抗作用。 因此,不能作为体外抗凝药使用,体内抗凝也须有活性的凝血因 子消耗后才能有效,起效后作用和维持时间亦较长。主要用于防 治血栓栓塞性疾病。
本药为香豆素类抗凝药之一,香豆素类抗凝药是一类含有4-羟基 香豆素基本结构的物质,口服参与体内代谢才发挥抗凝作用,故称口 服抗凝药。有双香豆素(dicoumarol)、华法林(warfarin,苄丙酮香 豆素)和醋硝香豆素(acenocoumarol,新抗凝)等。它们的药理作用 相同。
药理作用:香豆素类是维生素K拮抗剂,在肝脏抑制维生素K 由环氧化物向氢醌型转化,从而阻止维生素K的反复利用,影响含 有谷氨酸残基的凝血因子II、VII、IX、X的羧化作用,使这些因子停 留于无凝血活性的前体阶段,从而影响凝血过程。对已形成的上述因 子无抑制作用,因此抗凝作用出现时间较慢。一般需8~12小时后发 挥作用,1~3天达到高峰,停药后抗凝作用尚可维持数天。双香豆 素抗凝作用慢而持久,持续4~7天。华法林作用出现较快,持续2~ 5天。
体内过程:华法林口服吸收完全,1小时后血浆中即能测到,2~ 8小时达高峰。与血浆蛋白结合率为90%~99%。t1/2为10~60小时。 主要在肝及肾中代谢。双香豆素吸收不规则。与血浆蛋白结合率为 90%~99%。t1/2为10~30小时。醋硝香豆素t1/2为8小时,还原 型代谢产物仍有抗凝作用,t1/2为20小时。
临床应用:用途与肝素同,可防止血栓形成与发展。也可作为心 肌梗塞辅助用药。口服有效,作用时间较长。但作用出现缓慢,剂量 不易控制。也用于风湿性心脏病、髋关节固定术、人工置换心脏瓣膜 等手术后防止静脉血栓发生。
药物相互作用:①食物中维生素K缺乏或应用广谱抗生素抑制 肠道细菌,使体内维生素K含量降低,可使本类药物作用加强。② 阿司匹林等血小板抑制剂可与本类药物发生协同作用。③水合氯醛、 羟基保泰松、甲磺丁脲、奎尼丁等可因置换血浆蛋白,水杨酸盐、丙 咪嗪、甲硝唑、西咪替丁等因抑制肝药酶均使本类药物作用加强。④ 巴比妥类、苯妥英钠因诱导肝药酶,口服避孕药因增加凝血作用可使 本类药物作用减弱。
华法林抗凝治疗的适应症:
(一)瓣膜病和瓣膜置换:瓣膜病(尤其二尖瓣狭窄)如果合并 房颤或者已经发生了脑栓塞,或者超声心动图发现左房直径明显增大 (>55mm),应长期口服华法林,维持INR于2.0~3.0,否则口服阿 司匹林就可以了。强烈推荐所有机械瓣换瓣病人接受长期(永久)口 服抗凝药物治疗,一般维持INR于2.5~3.5。如果病人同时存在房颤 或者口服华法林抗凝过程中仍然发生了血栓栓塞,应考虑加用阿司匹 林。
生物瓣置换发生血栓栓塞的风险明显小于机械瓣,一般术后抗凝 3个月即可,维持INR于2.0~3.0。如果生物瓣置换病人同时合并房 颤或者手术时发现房内血栓,应长期口服华法林治疗;如果既往有血 栓栓塞病史,抗凝治疗的时间应在3~12个月。其他情况长期口服阿 司匹林即可。
(二)非瓣膜病性房颤:多个随机试验的荟萃分析显示,在非瓣 膜病性房颤病人,与安慰剂比较,华法林使脑卒中的危险下降68%, 大出血的发生率没有显著性增加,抗凝降低死亡率33%。阿司匹林 也有效,总体上缺血性脑卒中的危险下降21%,效果不及华法林。 在房颤已发生了脑栓塞的病人,口服抗凝剂降低年主要心血管事件 (血管性死亡、非致命性脑卒中、非致命性心肌梗死或周围血管栓塞) 47%,脑卒中危险下降66%,抗凝组没有病人发生脑出血。阿司匹 林只降低主要终点事件17%,脑卒中下降14%,与安慰剂对照组比 较无统计学差异。
年龄越大,血栓栓塞的风险也越大。年龄大于75岁的持续房颤, 应常规口服华法林抗凝治疗,华法林对于这类病人获益明显大于风 险。高龄病人对于华法林的敏感性增加,不但体现在出血的风险增加, 还表现在同样的INR水平,老年人获得的实际抗栓水平更高,因此 可以考虑将华法林剂量调整为INR1.6~2.5。
所有同时存在其他血栓危险因素的房颤病人都应接受华法林抗 栓治疗。这些危险因素包括先前的TIA、周围血管栓塞或脑卒中、高 血压、左室功能低下、风湿性二尖瓣病、瓣膜置换。其他包括较高龄、 明显的冠状动脉疾病、糖尿病、甲亢,超声发现左室功能不全、左房 增大、左房流速减慢、左房内自发回声。有脑卒中或短暂脑缺血发作 病史的病人,年脑卒中发生率可以达到12%。年龄65~75岁之间, 如果不存在其他危险因素,口服华法林或者阿司匹林都可以。年龄小 于65岁,又不存在引起血栓栓塞的其他危险因素,口服阿司匹林即 可。
强烈推荐有适应症的高危病人长期口服抗凝剂,维持目标INR 2.5(2.0~3.0),阿司匹林的效果不如抗凝剂。阵发性房颤发生血栓 栓塞的风险也是增加的,是否应用华法林应该主要取决于病人是否存 在引起血栓栓塞的其他危险因素。
(三)电复律:对于房颤大于48小时电复律的病人,复律前应 抗凝3周,维持目标INR 2.5(2.0~3.0),复律成功后抗凝4周,以 免因心耳部位收缩延迟恢复,形成新的血栓栓塞。据报道,口服奎尼 丁复律栓塞发生率在400个病人中为1.5%,因此建议药物复律同电 复律一样,需要口服抗凝药物预处理,然后复律,方法同房颤。
复律前华法林合并使用肝素或者低分子肝素可能会缩到复律前 用药的时间,但需要进一步探讨其效果。以食管超声确定复律前是否 需要口服华法林3周并不十分可靠。
(四)冠心病:对于冠心病的一级预防和二级预防,中等强度的 华法林(INR 2.5~3.5)预防血管事件的效果至少与阿司匹林相当。 如果中等剂量的华法林(INR 2.0~3.0)加阿司匹林,效果好于任何 一个药物单独使用,但两者合用有可能增加出血的风险。
(五)肺栓塞和深静脉血栓形成:一般的做法是,在肝素或低分 子肝素治疗的基础之上,24小时内同时给予华法林口服,待给肝素 4~5日以上或连续2日INR
1961年,West等(J.Am.Chem.Soc.1961,81,2676)曾用奎宁定, 奎宁的金属盐化合物成功的分离出单一立体构型的华法林(R和S)。 遗憾的是他的产率只有31%。Cook等 (J.PHarmcol.Exp.Ther.1979,210(3),391)用樟脑磺酰氯对华法林做进 一步的衍生,将得到的产物柱分离,而后用5%NaOH水溶液处理, 分别得到R,S单一构型的华法林,产率分别为10.5%、12.2%。Ohta等 (J.Org.Chem.1995,60.357-363)报道了一种采用有机膦做催化剂通过 不对称催化香豆素和一系列不饱和环烯酮来合成手性的类似结构的 化合物。但是遗憾的是当不饱和烯酮的取代基为苯基,即产物结构为 华法林时,并未取得很好的实验结果。
因此就目前的工艺而言,工业化生产单一构型是华法林及其衍生 物是很困难的事情,这就要求我们寻找一种更为简单、高效、有用的 合成工艺来应用于工业化生产R/S-华法林及其衍生物。
发明内容
本发明需要解决的技术问题是,基于现有生产工艺的不足,提供 了一种可替代现有路线的操作简便、成本较低的适合于工业化生产华 法林的合成路线。
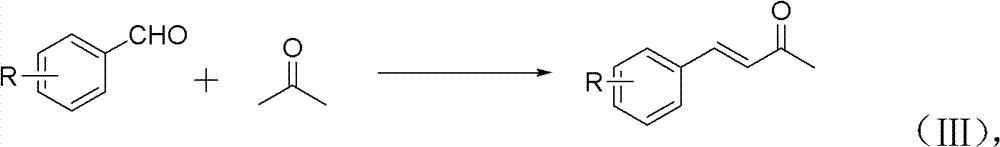
本发明中化合物(1)-(12)通过如下合成路线实施:先以含各 种取代基的苯甲醛和丙酮为原料合成中间产物各种取代的亚苄基丙 酮,再将中间产物与各种取代的4-羟基香豆素反应,得到最终产物华 法林及其衍生物。
具体地,本发明的手性华法林及其衍生物的制备方法,按如下步 骤:
1)在乙醇与水体积比为5∶2~3的混合溶剂中,取代的苯甲醛与 丙酮的投料摩尔比为1∶2,进行醇醛反应,反应温度为25~30℃, 时间为3~6h;反应过程中通过添加NaOH为重量5~15%的NaOH水 溶液保持碱性环境;反应结束后,经分离纯化(即萃取,水洗,干燥, 过滤,去溶剂,减压蒸馏,冷却),得淡黄色固体——中间产物取代 亚苄基丙酮;见反应式(III):
2)在反应溶剂中,4-羟基香豆素与取代亚苄基丙酮的投料摩尔 比为1∶1.5~1.6,在手性催化剂、相对于4-羟基香豆素1~10倍量的乙 酸、和辅助催化剂的共同作用下经1,4-加成反应,反应温度为25~ 30℃,时间为22~24h,反应结束后调节反应液的pH为中性,经后处 理得到手性华法林及其衍生物产品,见反应式(IV):
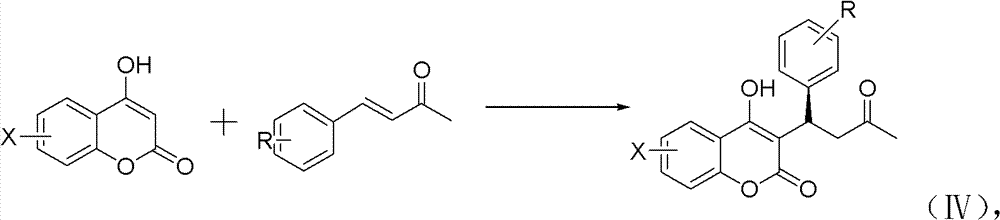
上式中R=H,CH3,OC H3,Cl或Br;X=H,Cl或Br;它们 分别对应如下结构的产品:
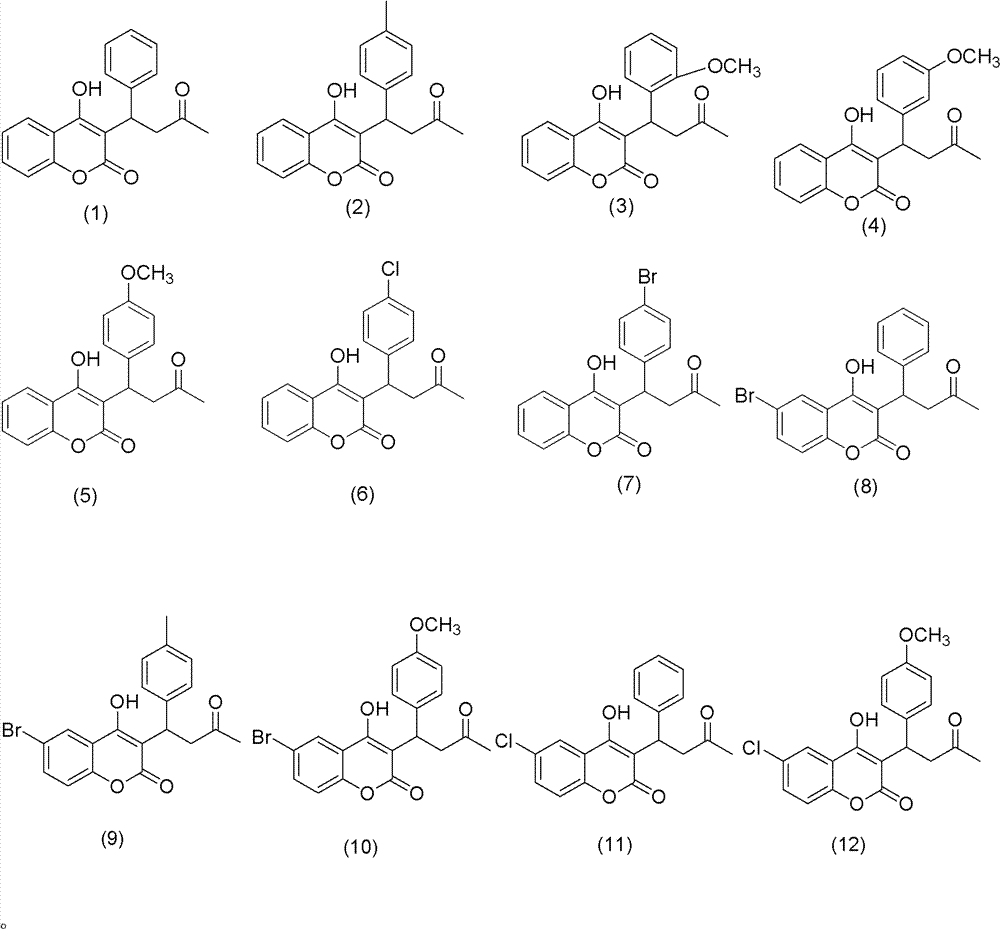
所述的手性催化剂为手性伯胺催化剂,选自(R,R)二苯基乙二 胺、(S,S)-二苯基乙二胺、(R,R)-环己二胺或(S,S)-环己二胺;
所述的辅助催化剂(为金属化合物)选自三氯化铁、高氯酸镁、 高氯酸锂、五氯化铌;
所述的反应溶剂选自1,4-二氧六环、四氢呋喃、乙醚、甲基叔丁 基醚等醚类溶剂。
与背景技术相比,本发明方法具有操作简便、成本较低、更适合 于工业化生产等优点。
附图说明
图1是实施例1消旋华法林的HPLC图;图2是消旋华法林的结 构式图。
图3是实施例1R-华法林的HPLC图;图4R-华法林的结构式 图。
具体实施方式
下面对本发明的实施例做详细的说明,本实施例在以发明技术方 案为前提进行实施,给出了详细的实施方式和具体的操作过程但本发 明的保护范围不限于下面的实施例。
实施例1
1)中间产物取代亚苄基丙酮的制备。反应式:
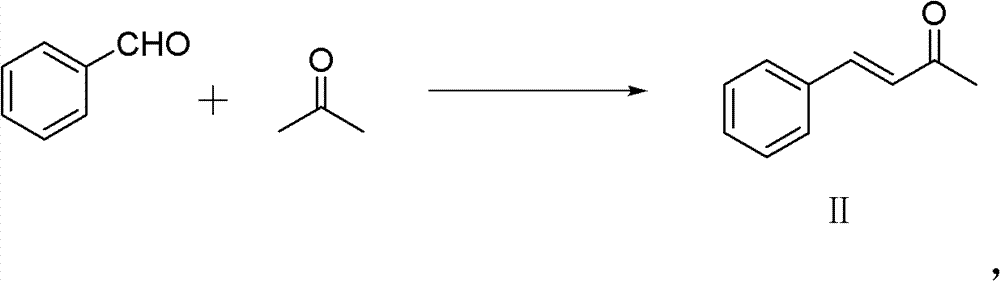
操作步骤:
50ml单口圆底烧瓶,加入苯甲醛(0.02mol)和丙酮(0.56mmol)。 再加入10%的NaOH水溶液0.5ml,再加入2ml H2O。室温下搅拌3h。 反应结束后用稀HCl调节pH使溶液呈中性。分离纯化(减压蒸馏), 得粘稠的油状液体,冷却后得淡黄色固体——亚苄基丙酮 (150~160℃/3333Pa(7mmHg))。
2)手性华法林的制备。反应式:
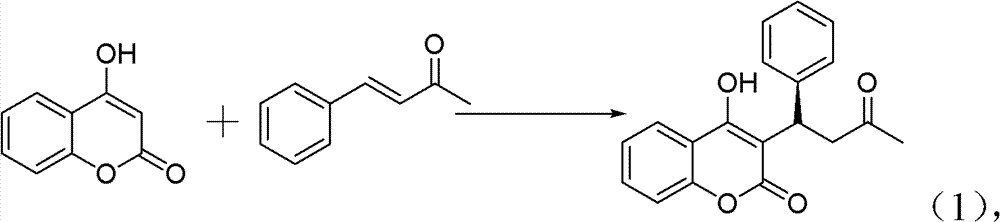
操作步骤:
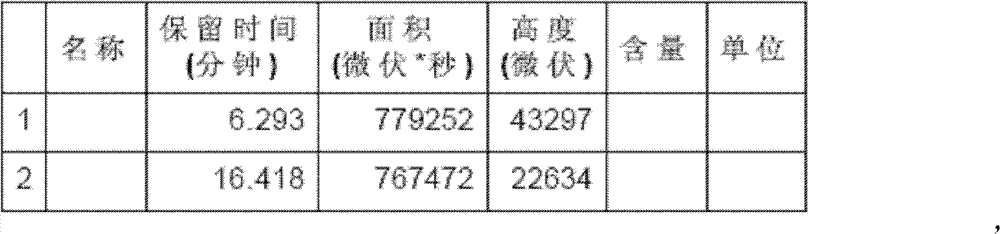
10ml的反应试管,一次性加入4-羟基香豆素(0.1m mol)、亚 苄基丙酮(0.12m mol)、手性伯胺催化剂(5m mol%)、高氯酸锂(5m mol%)、乙酸(1mmol,10eq),最后再加入1,4-二氧六环2ml。在室 温下搅拌24h,反应结束后加水淬灭。用CH2Cl2萃取(10ml×3次), 有机相用水洗三次(10ml),无水硫酸钠干燥有机相。旋干溶剂过柱 子(石油醚∶乙酸乙酯=5∶1)得白色固体——式(1)手性华法林。 见图1、图2,其中

峰结果
和图3、图4,其中

峰结果

其化合物表征数据:
1H-NMR(400MHz,CDCl3):δ(ppm)=1.68(s,1.60H,CH3,ketal, 1.72(s,1.37H,CH3,ketal),1.97-2.04(t,0.67H,CH2,ketal),2.30(s, 0.46H,CH3,ketal),2.40-2.60(m,1.65H,CH2,keto),3.30-3.34(d,J= 19.2Hz,0.29H,CH2,ketal),3.83-3.39(dd,J=19.2Hz,0.22H,CH2, keto),4.14-4.19(m,0.54H,CH,ketal),4.28-4.30(m,0.43H,CH ketal), 4.70-4.72(d,J=9.6Hz,0.20H,CH,keto),7.23-7.30(m,7.37H,ArH), 7.50(t,0.72H,ArH),7.80,7.82(d,J=7.6Hz,0.45H),7.90,7.91(d,J= 8Hz,0.41Hz,7.94-7.96(d,J=7.6Hz,0.16H,ArH),9.50(S,0.21H,OH keto).13C-NMR(100MHz,CDCl3):δ(ppm)=212.318,171.166, 162.151,161.384,159.712,158.897,153.023,152.902,143.287,141.410, 132.04,131.60,129.27,128.65,128.19,127.99,127.26,127.05,126.97, 126.53,123.95,123.65,123.07,122.72,116.69,116.56,104.23,101.13, 100.50,98.96,42.51,39.97,35.35,34.13,28.28,27.78.IR(film): v=3277.7,1681.5,1617.7,1517.2,1496.2,1451.9,1377.4,1076.0,995.9, 763.4,700.1.GC m/z:309(M+),265(bp),187,120.HR S(ESI):calad. for C19H16O4H+:[M+H]309.1121;found 309.1125.HPLC(Daicel AD-H; 2-propanol/n-Hexane,20:80;flow rate=1.0mL/min;λ=254nm): tmajor=6.3min(major),tminor=16.4min(minor).
实施例2
1)中间产物4-甲基亚苄基丙酮的制备,反应式:
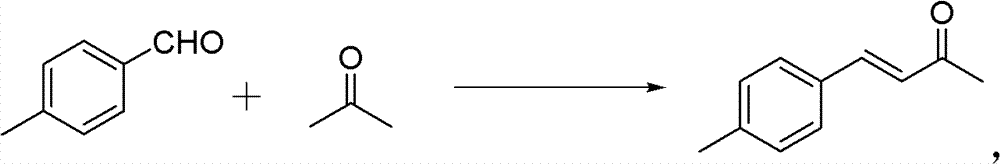
操作步骤:
50ml单口圆底烧瓶,加入4-甲基苯甲醛(0.02mol)和丙酮 (0.56mmol)。再加入10%的NaOH水溶液0.5ml,再加入2ml H2O。 室温下搅拌3h。反应结束后用稀HCl调节pH使溶液呈中性。分离纯 化(减压蒸馏),得粘稠的油状液体,冷却后得淡黄色固体——亚苄 基丙酮(150-160℃/3333Pa(7mmHg))。
2)手性华法林衍生物的制备,反应式:
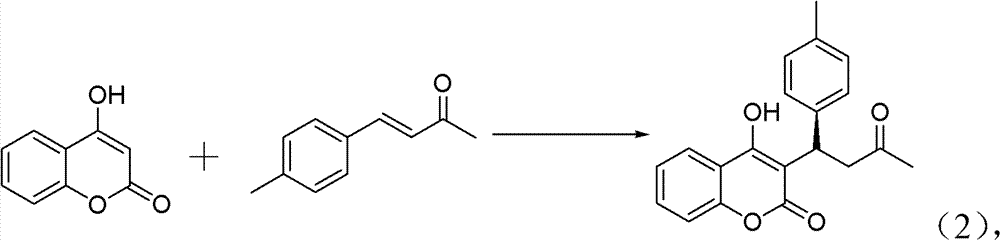
操作步骤:
10ml的反应试管,一次性加入4-羟基香豆素(0.1621g,0.1mmol, 1eq)、中间产物4-甲基亚苄基丙酮(0.220g,0.12mmol,1.2eq)、手 性伯胺催化剂(5mmol%)、高氯酸锂(5mmol%)、乙酸(1mmol,10eq), 最后再加入1,4-二氧六环2ml。在室温下搅拌24h,反应结束后加水 淬灭。用CH2Cl2萃取(10ml×3次),有机相用水洗三次(10ml),无 水硫酸钠干燥有机相。旋干溶剂过柱子(石油醚∶乙酸乙酯=5∶1) 得白色固体——式(2)手性华法林衍生物3-(3-氧代-1-对甲基苯基丁 基)-4-羟基-2H-1-苯并吡喃-2-酮。
实施例3
1)中间产物2-甲氧基亚苄基丙酮的制备。反应式:

操作步骤:
50ml单口圆底烧瓶,加入2-甲氧基苯甲醛(0.02mol)和丙酮 (0.56mmol)。再加入10%的NaOH水溶液0.5ml,再加入2ml H2O。 室温下搅拌3h。反应结束后用稀HCl调节pH使溶液呈中性。分离纯 化(减压蒸馏)的馏分,得粘稠的油状液体,冷却后得淡黄色固体— —亚苄基丙酮(150-160℃/3333Pa(7mmHg))
2)手性华法林衍生物的制备,反应式:
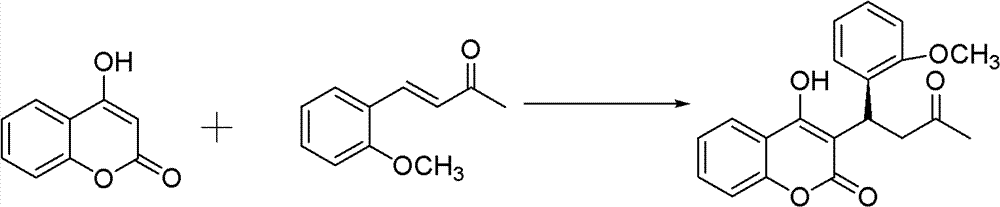
(3);
操作步骤:
10ml的反应试管,一次性加入4-羟基香豆素(0.1mmol)、中间 产物2-甲氧基亚苄基丙酮(0.12mmol)、手性伯胺催化剂(5mmol%)、 高氯酸锂(5mmol%)、乙酸(1mmol,10eq),最后再加入1,4-二氧六 环2ml。在室温下搅拌24h,反应结束后加水淬灭。用CH2Cl2萃取 (10ml×3次),有机相用水洗三次(10ml),无水硫酸钠干燥有机相。 旋干溶剂过柱子(石油醚∶乙酸乙酯=5∶1)得白色固体——式(3) 手性华法林衍生物3-(3-氧代-1-2’-甲氧基苯基丁基)-4-羟基-2H-1-苯并 吡喃-2-酮。
实施例4
1)中间产物3-甲氧基亚苄基丙酮的制备。反应式:
操作步骤:
50ml单口圆底烧瓶,加入3-甲氧基苯甲醛(0.02mol)和丙酮 (0.56mmol)。再加入10%的NaOH水溶液0.5ml,再加入2ml H2O。 室温下搅拌3h。反应结束后用稀HCl调节pH使溶液呈中性。反应结 束后分离纯化(减压蒸馏),得粘稠的油状液体,冷却后得淡黄色固 体——亚苄基丙酮(150-160℃/3333Pa(7mmHg))。
2)手性华法林衍生物的制备,反应式:
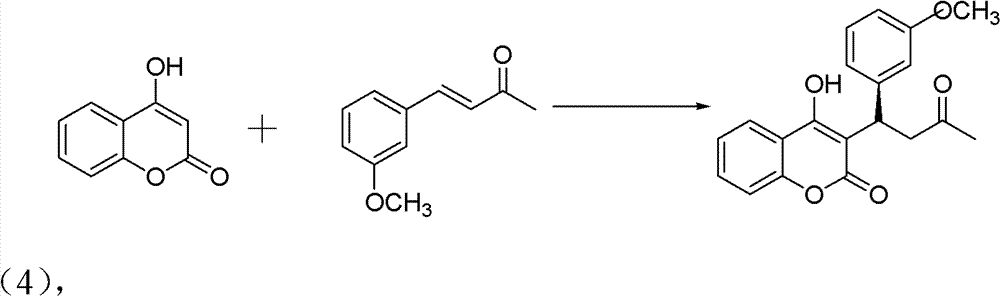
操作步骤:
10ml的反应试管,一次性加入4-羟基香豆素(0.1mmol)、中间 产物3-甲氧基亚苄基丙酮(0.12mmol)、手性伯胺催化剂(5mmol%)、 高氯酸锂(5mmol%)、乙酸(1mmol,10eq),最后再加入1,4-二氧六 环2ml。在室温下搅拌24h,反应结束后加水淬灭。用CH2Cl2萃取 (10ml×3次),有机相用水洗三次(10ml),无水硫酸钠干燥有机相。 旋干溶剂过柱子(石油醚∶乙酸乙酯=5∶1)得白色固体——式(4) 手性华法林衍生物3-(3-氧代-1-3’-甲氧基苯基丁基)-4-羟基-2H-1-苯并 吡喃-2-酮。
实施例5
1)中间产物4-甲氧基亚苄基丙酮的制备。反应式:
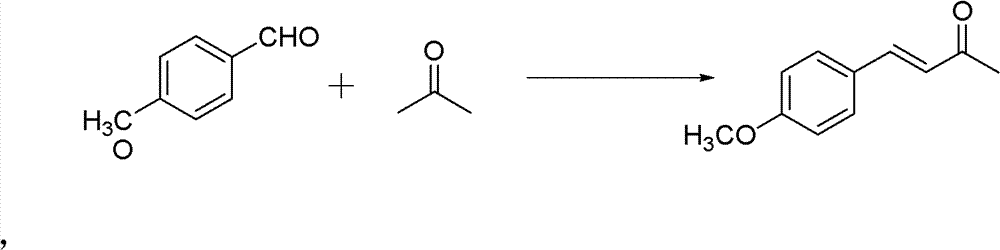
操作步骤:
50ml单口圆底烧瓶,加入4-甲氧基苯甲醛(0.02mol)和丙酮 (0.56mmol)。再加入10%的NaOH水溶液0.5ml,再加入2ml H2O。 室温下搅拌3h。反应结束后用稀HCl调节pH使溶液呈中性。萃取、 水洗、盐洗(饱和钠)、干燥(无水硫酸镁)、过滤除去干燥剂,旋干 溶剂过柱子(石油醚∶乙酸乙酯=12∶1)得淡黄色固体。
2)手性华法林衍生物的制备,反应式:
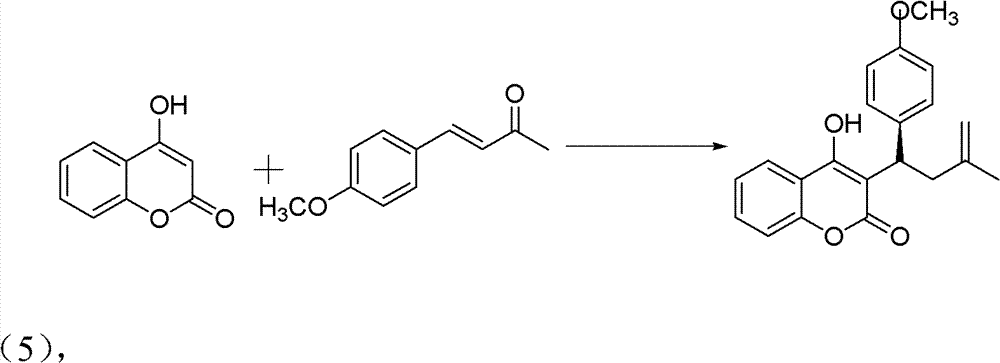
操作步骤:
10ml的反应试管,一次性加入4-羟基香豆素(0.1mmol)、中间 产物4-甲氧基亚苄基丙酮(0.12mmol)、手性伯胺催化剂(5mmol%)、 高氯酸锂(5mmol%)、乙酸(1mmol,10eq),最后再加入1,4-二氧六 环2ml。在室温下搅拌24h,反应结束后加水淬灭。用CH2Cl2萃取 (10ml×3次),有机相用水洗三次(10ml),无水硫酸钠干燥有机相。 旋干溶剂过柱子(石油醚∶乙酸乙酯=5∶1)得白色固体——式(5) 手性华法林衍生物3-(3-氧代-1对甲氧基苯基丁基)-4-羟基-2H-1-苯并 吡喃-2-酮。。
实施例6
1)中间产物4-氯亚苄基丙酮的制备。反应式:

(6),
操作步骤:
50ml单口圆底烧瓶,加入4-氯苯甲醛(0.02mol)和丙酮 (0.56mmol)。再加入10%的NaOH水溶液0.5ml,再加入2ml H2O。 室温下搅拌3h。反应结束后用稀HCl调节pH使溶液呈中性。将反应 混合物转入分液漏斗中,分出有机层,水层用10ml甲苯萃取,合并 有机相和甲苯萃取液,用10ml水洗涤后,用无水硫酸镁干燥。过滤 除去干燥剂,旋干溶剂过柱子(石油醚∶乙酸乙酯=10∶1)得淡黄色 固体。
2)手性华法林衍生物的制备,反应式:
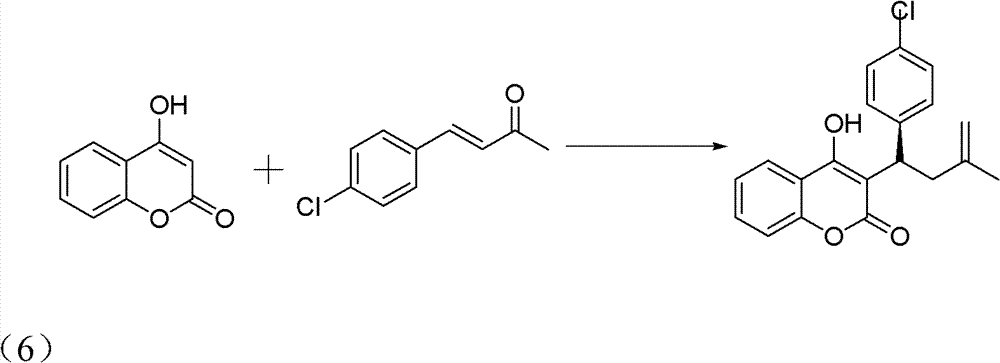
操作步骤:
10ml的反应试管,一次性加入4-羟基香豆素(0.1mmol)、中间 产物4-氯亚苄基丙酮(0.12mmol)、手性伯胺催化剂(5mmol%)、高 氯酸锂(5mmol%)、乙酸(1mmol,10eq),最后再加入1,4-二氧六环 2ml。在室温下搅拌24h,反应结束后加水淬灭。用CH2Cl2萃取(10ml×3 次),有机相用水洗三次(10ml),无水硫酸钠干燥有机相。旋干溶剂 过柱子(石油醚∶乙酸乙酯=5∶1)得白色固体——式(6)手性华法 林衍生物3-(3-氧代-1-对氯苯基丁基)-4-羟基-2H-1-苯并吡喃-2-酮。
实施例7
1)中间产物4-溴亚苄基丙酮的制备。反应式:

操作步骤:
50ml单口圆底烧瓶,加入4-溴苯甲醛(0.02mol)和丙酮 (0.56mmol)。再加入10%的NaOH水溶液0.5ml,再加入2ml H2O。 室温下搅拌3h。反应结束后用稀HCl调节pH使溶液呈中性。将反应 混合物转入分液漏斗中,分出有机层,水层用10ml甲苯萃取,合并 有机相和甲苯萃取液,用10ml水洗涤后,用无水硫酸镁干燥。过滤 除去干燥剂,旋干溶剂过柱子(石油醚∶乙酸乙酯=10∶1)得淡黄色 固体。
2)手性华法林衍生物的制备,反应式:
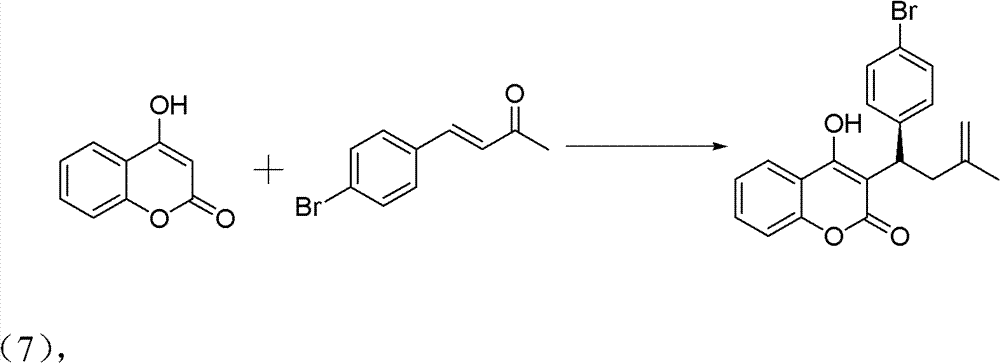
操作步骤:
10ml的反应试管,一次性加入4-羟基香豆素(0.1mmol)、中间 产物2-溴亚苄基丙酮(0.12mmolq)、手性伯胺催化剂(5mmol%)、 高氯酸锂(5mmol%)、乙酸(1mmol,10eq),最后再加入溶剂2ml。 在室温下搅拌24h,反应结束后加水淬灭。用CH2Cl2萃取(10ml×3 次),有机相用水洗三次(10ml),无水硫酸钠干燥有机相。旋干溶剂 过柱子(石油醚∶乙酸乙酯=5∶1)得白色固体——式(7)手性华法 林衍生物3-(3-氧代-1对溴基苯基丁基)-4-羟基-2H-1-苯并吡喃-2-酮。。
实施例8
1)中间产物亚苄基丙酮的制备。反应式:
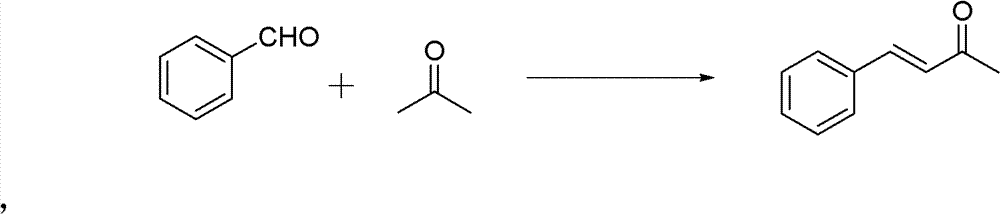
操作步骤:
50ml单口圆底烧瓶,加入苯甲醛(0.02mol)和丙酮(0.56mmol)。 再加入10%的NaOH水溶液0.5ml,再加入2ml H2O。室温下搅拌3h。 反应结束后用稀HCl调节pH使溶液呈中性。分离纯化(减压蒸馏), 得粘稠的油状液体,冷却后得淡黄色固体——亚苄基丙酮 (150-160℃/3333Pa(7mmHg))。
2)手性华法林衍生物的制备,反应式:
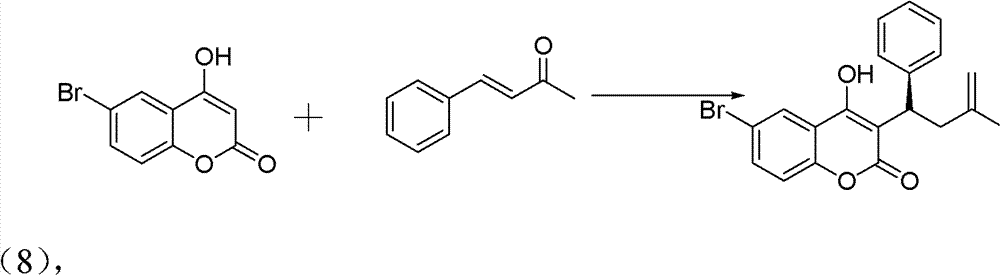
操作步骤:
10ml的反应试管,一次性加入6-溴-4-羟基香豆素(0.1mmol)、 中间产物亚苄基丙酮(0.12mmol)、手性伯胺催化剂(5mmol%)、高 氯酸锂(5mmol%)、乙酸(1mmol,10eq),最后再加入1,4-二氧六环 2ml。在室温下搅拌24h,反应结束后加水淬灭。用CH2Cl2萃取(10ml×3 次),有机相用水洗三次(10ml),无水硫酸钠干燥有机相。旋干溶剂 过柱子(石油醚∶乙酸乙酯=5∶1)得白色固体——式(8)手性华法 林衍生物3-(3-氧代-1苯基丁基)-4-羟基-2H-6-溴-1-苯并吡喃-2-酮。
实施例9
1)中间产物4-甲基亚苄基丙酮的制备。反应式:
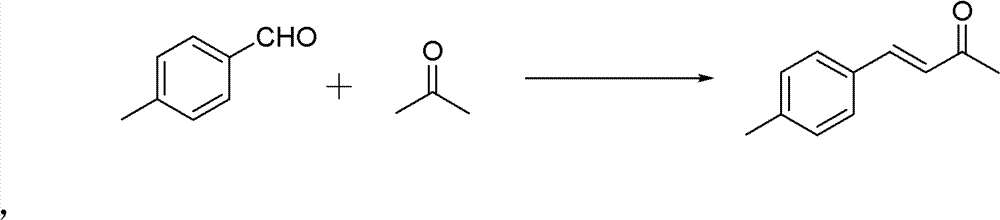
操作步骤:
50ml单口圆底烧瓶,加入4-甲基苯甲醛(0.02mol)和丙酮 (0.56mmol)。再加入10%的NaOH水溶液0.5ml,再加入2ml H2O。 室温下搅拌3h。反应结束后用稀HCl调节pH使溶液呈中性。分离纯 化(减压蒸馏),得粘稠的油状液体,冷却后得淡黄色固体——亚苄 基丙酮(150-160℃/3333Pa(7mmHg))
2)手性华法林衍生物的制备,反应式:
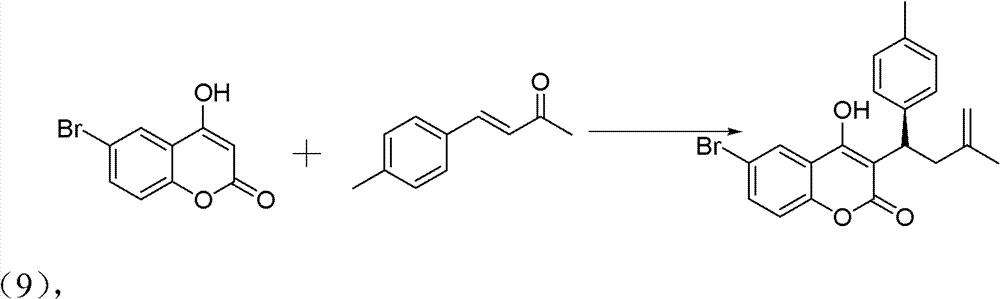
操作步骤:
10ml的反应试管,一次性加入6-溴-4-羟基香豆素(0.1mmol)、 中间产物4-甲基亚苄基丙酮(0.12mmol)、手性伯胺催化剂 (5mmol%)、高氯酸锂(5mmol%)、乙酸(1mmol,10eq),最后再加 入1,4-二氧六环2ml。在室温下搅拌24h,反应结束后加水淬灭。用 CH2Cl2萃取(10ml×3次),有机相用水洗三次(10ml),无水硫酸钠 干燥有机相。旋干溶剂过柱子(石油醚∶乙酸乙酯=5∶1)得白色固 体——式(9)手性华法林衍生物3-(3-氧代-1对甲基苯基丁基)-4-羟 基-2H-6-溴-1-苯并吡喃-2-酮。
实施例10
1)中间产物4-甲氧基亚苄基丙酮的制备。反应式:
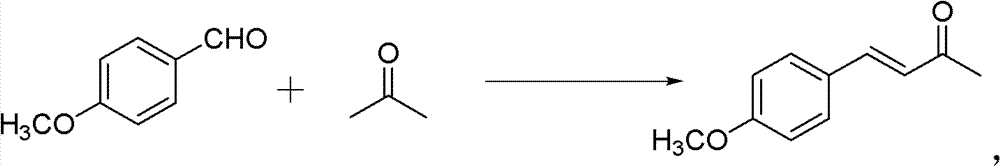
操作步骤:
50ml单口圆底烧瓶,加入4-甲氧基苯甲醛(0.02mol)和丙酮 (0.56mmol)。再加入10%的NaOH水溶液0.5ml,再加入2ml H2O。 室温下搅拌3h。反应结束后用稀HCl调节pH使溶液呈中性。分离纯 化(减压蒸馏),得粘稠的油状液体,冷却后得淡黄色固体——亚苄 基丙酮(150-160℃/3333Pa(7mmHg))。
2)手性华法林衍生物的制备,反应式:
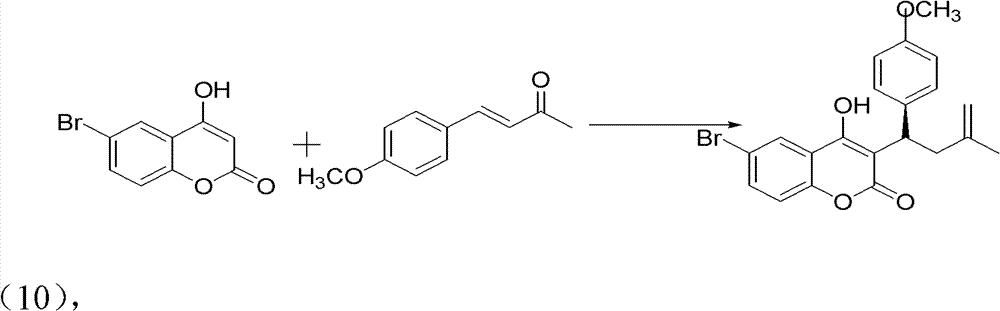
操作步骤:
10ml的反应试管,一次性加入6-溴-4-羟基香豆素(0.1mmol)、 中间产物4-甲氧基亚苄基丙酮(0.12mmol)、手性伯胺催化剂 (5mmol%)、高氯酸锂(5mmol%)、乙酸(1mmol,10eq),最后再加 入1,4-二氧六环2ml。在室温下搅拌24h,反应结束后加水淬灭。用 CH2Cl2萃取(10ml×3次),有机相用水洗三次(10ml),无水硫酸钠 干燥有机相。旋干溶剂过柱子(石油醚∶乙酸乙酯=5∶1)得白色固 体——式(10)手性华法林衍生物3-(3-氧代-1对甲氧基苯基丁基)-4- 羟基-2H-6-溴-1-苯并吡喃-2-酮。
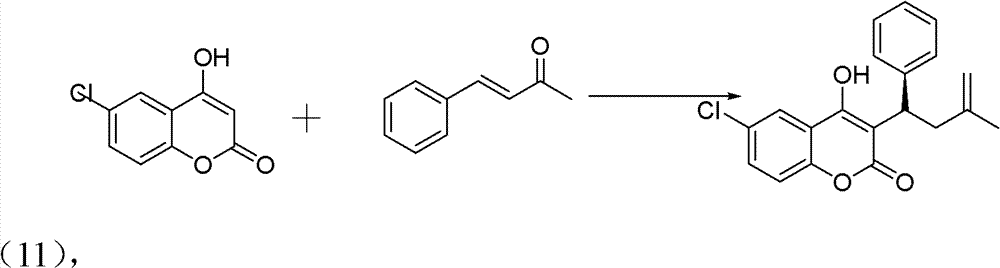
实施例11
1)中间产物亚苄基丙酮的制备。反应式:
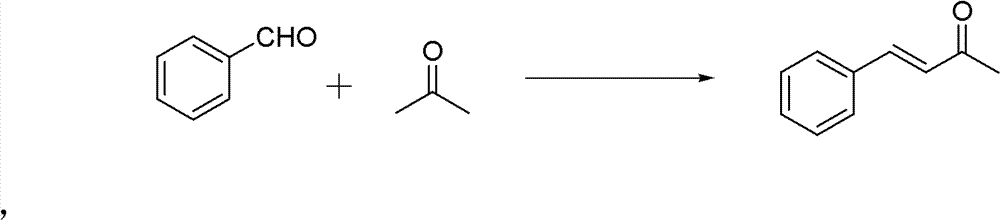
操作步骤:
50ml单口圆底烧瓶,加入苯甲醛(0.02mol)和丙酮(0.56mmol)。 再加入10%的NaOH水溶液0.5ml,再加入2ml H2O。室温下搅拌3h。 反应结束后用稀HCl调节pH使溶液呈中性。分离纯化(减压蒸馏), 得粘稠的油状液体,冷却后得淡黄色固体——亚苄基丙酮 (150-160℃/3333Pa(7mmHg))。
2)手性华法林衍生物的制备,反应式:
操作步骤:
10ml的反应试管,一次性加入6-氯-4-羟基香豆素(0.1mmol)、 中间产物亚苄基丙酮(0.12mmol)、手性伯胺催化剂(5mmol%)、高 氯酸锂(5mmol%)、乙酸(1mmol,10eq),最后再加入1,4-二氧六环 2ml。在室温下搅拌24h,反应结束后加水淬灭。用CH2Cl2萃取(10ml×3 次),有机相用水洗三次(10ml),无水硫酸钠干燥有机相。旋干溶剂 过柱子(石油醚∶乙酸乙酯=5∶1)得白色固体——式(11)手性华 法林衍生物3-(3-氧代-1苯基丁基)-4-羟基-2H-6-氯-1-苯并吡喃-2-酮。
实施例12
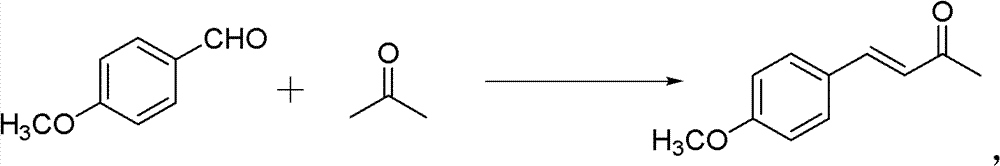
1)中间产物4-甲氧基亚苄基丙酮的制备。反应式:
操作步骤:
50ml单口圆底烧瓶,加入4-甲氧基苯甲醛(0.02mol)和丙酮 (0.56mmol)。再加入10%的NaOH水溶液0.5ml,再加入2ml H2O。 室温下搅拌3h。反应结束后用稀HCl调节pH使溶液呈中性。将反应 混合物转入分液漏斗中,分出有机层,水层用10ml甲苯萃取,合并 有机相和甲苯萃取液,用10ml水洗涤后,用无水硫酸镁干燥。旋干 溶剂过柱子(石油醚∶乙酸乙酯=12∶1)得淡黄色固体。
2)手性华法林衍生物的制备,反应式:
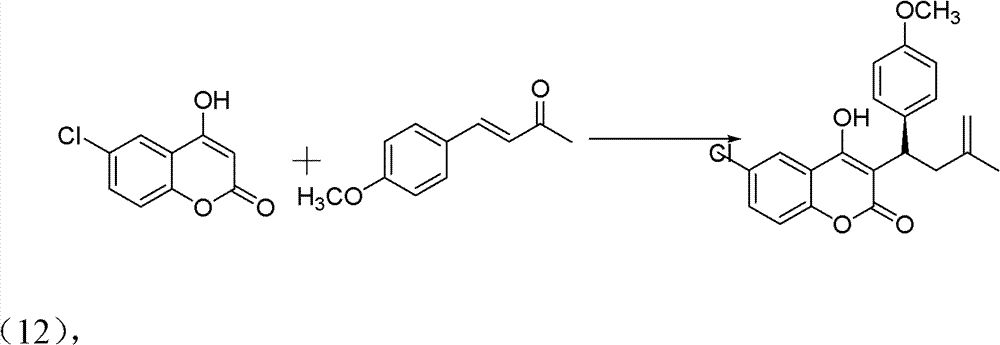
操作步骤:
10ml的反应试管,一次性加入6-氯-4-羟基香豆素(0.1mmol)、 中间产物4-甲氧基亚苄基丙酮(0.12mmol)、手性伯胺催化剂 (5mmol%)、高氯酸锂(5mmol%)、乙酸(1mmol,10eq),最后再加 入1,4-二氧六环2ml。在室温下搅拌24h,反应结束后加水淬灭。用 CH2Cl2萃取(10ml×3次),有机相用水洗三次(10ml),无水硫酸 钠干燥有机相。旋干溶剂过柱子(石油醚∶乙酸乙酯=5∶1)得白色 固体——式(12)手性华法林衍生物3-(3-氧代-1-3’-甲氧基苯基丁 基)-4-羟基-2H-6-氯-1-苯并吡喃-2-酮。

 手机二维码
手机二维码 京公网安备11011302007134
京公网安备11011302007134