(一)技术领域
本发明涉及一种α-硫氰化酮化合物的合成方法。
(二)背景技术
α-硫氰化酮作为一类重要的化合物,广泛应用于农药、医药以及众多 精细化学品的合成,同时在某些天然抗癌药物的制备中也有较好的应用前 景[Metzer,J.B.Comprehensive Heterocyclic Chemistry;Katritzky,A.,Ed.; Pergamon:Oxford,1984.];[Shahidi,F.Sulphur Compounds in Foods; Mussinan,C.J.,Keelan,M.E.,Eds.;American Chemical Society: Washington,DC,1984.]。因此,发展简便、高效、具有广泛适用性的制备 α-硫氰化酮的方法具有非常重要的应用价值。
目前,合成α-硫氰化酮的主要方法是通过酮α位的离去基团(如Br、 Cl、OTs等)与SCN-(如KSCN,NaSCN,Zn(SCN)2,NH4SCN等)经亲核 取代反应而得[Dotsenko,V.V.;Krivokolysko,S.G.;Chernega,A.N.; Litvinov,V.P.Russ.Chem.Bull.2007,56,1431.]、[Bisogno,F.R.;Cuetos, A.;Lavandera,I.;Gotor,V.Green Chem.2009,11,452.]。但该法存在着反应 步骤较多、反应条件较苛刻等缺点。同时,由于硫氰根离子亲核性较弱, 故反应收率往往较低,在某种程度上限制了其工业化应用。
酮的直接氧化硫氰化是近年来发展起来的构建C-S键的一类重要方 法,它主要通过氧化剂的氧化过程来促进硫氰酸盐与酮的直接氧化偶联, 由于省去了酮的卤化步骤,故具有反应步骤少、总收率高等特点。目前已 报道的氧化硫氰化体系主要有Me3SiNCS/Br2[Tanabe,Y.;Makita,T.;Mori, K.Chem.Lett.1994,12,2275.];PhICl2/Pb(SCN)2[:Prakash,O.;Kaur,H.; Batra,H.;Rani,N.;Singh,S.P.;Moriarty,R.M.J.Org.Chem.2001,66, 2019.];I2/NH4SCN[Yadav,J.S.;Reddy,B.V.S.;Reddy,U.V.S.;Krishna,A. D.Tetrahedron Lett.2007,48,5243.];FeCl3/NH4SCN[Yadav,J.S.;Reddy,B. V.S.;Reddy,U.V.S.;Chary,D.N.Synthesis 2008,8,1283.]; Oxone/NH4SCN[Kumar,M.A.;Reddy,K.R.K.K.;Reddy,M.V.;Reddy,C. S.;Reddy,C.D.Synth.Commun.2008,38,2089.];Me2BrS+S-/NH4SCN [Bhalerao.D.S.;Akamanchi,K.G.Synth.Commun.2010,40,799.]等。但这 些体系中往往需要添加化学计量或超过化学计量的氧化剂以促进反应的 进行,这无疑大大增加了反应成本,同时这些氧化剂的还原副产物也会对 环境造成一定的危害。
(三)发明内容
本发明目的是提供一种α-硫氰化酮化合物的合成方法,该方法反应 条件温和、成本低廉、对环境无污染,产物产率高。
本发明采用的技术方案是:
一种α-硫氰化酮化合物的合成方法,所述的方法以式(I)~(III) 之一所示的酮和硫氰酸盐为原料,以氧气或空气为氧化剂,以氢溴酸或溴 为助催剂,在催化剂的作用下,于溶剂中,0~100℃反应1~48h,反应结 束,反应液后处理制得所述的α-硫氰化酮化合物;所述的催化剂为铜盐; 所述的溶剂为C1~C3醇、C1~C3酸或二甲基亚砜或水;所述的酮为芳香 酮或脂肪酮;
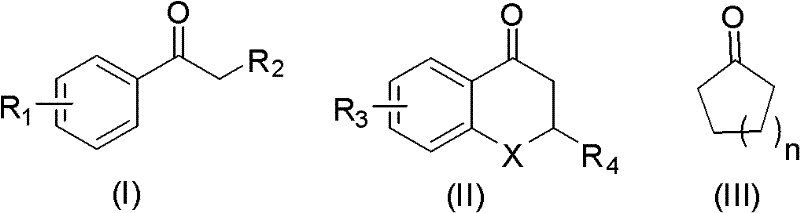
式(I)中R1为氢、C1~C4直链或支链烷烃基、C1~C3烷氧基、硝基、 卤素或取代甲基,所述的取代甲基的取代基为卤素;
R2为氢、C1~C3直链或支链烷烃、C1~C3烷氧基、二甲胺基或乙酰 基;
式(II)中R3为氢、C1~C3直链或支链烷烃基、C1~C3烷氧基;X 为CH2、O或NH;R4为氢或者苯基;
式(III)中n为1~4的自然数。
进一步,所述的酮优选为:式(I)中R1为氢、甲基、甲氧基、乙基、 丙基、异丙基、叔丁基、硝基、三氟甲基、氯、溴、氟,R1在苯基的2、 3或4位均可;
R2为氢、甲基、乙基、甲氧基、二甲胺基或乙酰基;
式(II)中R3为氢、甲基、乙基、甲氧基,R3在苯基的5、6、7或8 位均可;X为CH2、O或NH;R4为氢或者苯基;
式(III)中n为1~4的自然数。
更进一步,所述的酮优选对溴苯乙酮、α-四氢萘酮、环戊酮、邻甲基 苯乙酮、2-苯基色满-4-酮、环己酮、邻甲氧基苯乙酮或环庚酮。
所述的硫氰酸盐优选为下列之一:硫氰酸钠、硫氰酸钾、硫氰酸铵或 硫氰酸锌。
所述的催化剂优选为下列之一:碘化亚铜、氯化亚铜、溴化亚铜、溴 化铜、氯化铜、醋酸铜或三氟醋酸铜。
所述的溶剂为下列一种或两种以上任意比例的混合:水、甲醇、乙醇、 乙酸、丙酸或二甲基亚砜。
所述的α-硫氰化酮化合物的合成方法,所述的酮与硫氰酸盐、助催 剂、催化剂的投料物质的量之比为1∶1.2~3.0∶0.05~0.4∶0.1~0.6,优选为 1∶1.5~2.0∶0.1~0.25∶0.2~0.4。
所述的溶剂的体积用量以酮质量计为10~100mL/g。
所述的α-硫氰化酮化合物的合成方法,所述的后处理方法为:反应结 束,反应液加水稀释,用二氯甲烷萃取,水相用二氯甲烷萃取两次,合并 所有有机相,用饱和食盐水洗涤,再用无水硫酸钠干燥,过滤,滤液过层 析柱分离,以体积比1∶0.02~0.2的正己烷和乙酸乙酯混合液为洗脱剂, 制得α-硫氰化酮化合物;所述水的添加是用于萃取稀释用,水的添加量 多少对本发明没有影响,一般推荐所述反应液与水的体积比为1∶0.5~5。
所述的α-硫氰化酮化合物的合成方法,所述的方法推荐按照以下步 骤进行:在密闭的氧气或空气气氛中,将酮、硫氰酸盐、催化剂和助催剂 于溶剂中混合,在压力为0.1~0.5MPa下,于40~80℃反应5~24h,反应 结束,反应液用水稀释,再用二氯甲烷萃取,水相用二氯甲烷萃取两次, 合并所有有机相,用饱和食盐水洗涤,再用无水硫酸钠干燥,过滤,滤液 过层析柱分离,以体积比1∶0.02~0.2的正己烷和乙酸乙酯混合液为洗脱 剂,制得α-硫氰化酮化合物;所述的酮为对溴苯乙酮、α-四氢萘酮、环戊 酮、邻甲基苯乙酮、2-苯基色满-4-酮、环己酮、邻甲氧基苯乙酮、环庚酮; 所述的硫氰酸盐为硫氰酸钾、硫氰酸铵、硫氰酸锌或硫氰酸钠;所述的催 化剂为碘化亚铜、氯化亚铜、溴化亚铜、氯化铜、溴化铜、醋酸铜或三氟 醋酸酮;所述的助催剂为氢溴酸或溴;所述的溶剂为乙酸、甲醇、水、二 甲基亚砜或丙酸;所述的酮与硫氰酸盐、助催剂、催化剂的投料物质的量 之比为1∶1.5~2.0∶0.1~0.25∶0.2~0.4;所述的溶剂的体积用量以酮质量计 为10~100mL/g。
所述的α-硫氰化酮化合物的合成方法,反应过程在一个密闭的氧气 或者空气气氛中进行,催化剂(铜盐)和助催化剂(氢溴酸或溴素)的用量可 以变化,通常为催化量即可。
与现有技术相比,本发明的有益效果主要体现在:
(1)反应条件温和,操作易于控制;本发明在0~100℃下即可较好 地进行氧化硫氰化反应,条件温和;通常活性较好的芳香酮化合物在12h 内可得到收率较高的硫氰化产物,而脂环酮化合物反应时间相对较长;
(2)成本低廉,反应安全;同文献报道的传统氧化硫氰化体系相比, 本发明所提供的方法采用氧气或空气作氧化剂,铜盐作催化剂,氢溴酸或 溴素作助催化剂,原料价廉易得,具有成本低廉、使用安全等优点;
(3)整个过程对环境友好,无污染;传统的氧化硫氰化法通常需向 反应体系中加入化学计量或超过化学计量的氧化剂,从而大大增加了反应 成本,同时,这些氧化剂的还原副产物,不仅后处理麻烦,且对环境也将 造成一定的危害,而本发明所提供的氧化硫氰化方法,使用氧气或空气作 氧化剂,由于其还原副产物仅仅是水,从本质上减少了对环境的污染,因 此是一种绿色清洁的合成方法。
(四)具体实施方式
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围 并不仅限于此:
实施例1
在50mL配有磁子搅拌的Schlenk管中通过反复抽放氧气,使整 个体系处于全氧气气氛,然后向体系中加入对溴苯乙酮(I-1)0.196 g(1mmol),碘化亚铜0.038g(0.2mmol),氢溴酸(HBr)0.006mL(0.1 mmol),硫氰酸钾0.194g(1.5mmol),乙酸5mL,然后立即将整个体 系密闭在氧气气氛中,压力为0.1MPa,40℃下反应5h,反应结束, 反应液用5mL水稀释后,用10mL二氯甲烷萃取,水相继续用二氯 甲烷萃取两次(每次5mL),合并所有有机相,用饱和的食盐水洗涤, 再用无水硫酸钠干燥,过滤,滤液过层析柱,以体积比1∶0.04的正 己烷和乙酸乙酯混合液洗脱,得到产物α-硫氰化对溴苯乙酮0.205g, 收率为80%(以对溴苯乙酮物质的量计,下同)。
1H NMR(300MHz,CDCl3):4.69(s,2H),7.53(d,J=6.3Hz,2H), 7.88(d,J=6.3Hz,2H)。
I-1结构式: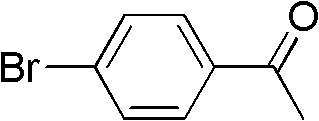
实施例2
芳香酮为α-四氢萘酮(II-1)0.146g(1mmol),催化剂为0.054g (0.3mmol)醋酸铜,助催剂为0.011mL(0.2mmol)HBr,硫氰酸盐 为0.145g(1.5mmol)硫氰酸钾,溶剂为甲醇5mL,压力为0.2MPa, 反应温度为55℃,反应时间为10h,其他操作同实施例1,得到硫氰 化四氢萘酮0.146g,率72%。
1H NMR(300MHz,CDCI3):d=8.0-8.03(m,1H),7.52-7.57 (m,1H),7.25-7.39(m,2H),4.54(dd,J=4.5,5.2Hz,1H),3.17-3.20(m, 2H),2.84-2.88(m,1H),2.35-2.44(m,1H).
II-1结构式: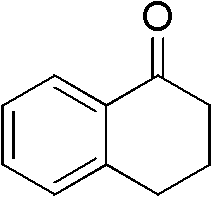
实施例3
脂肪酮为环戊酮(III-1)0.084g(1mmol),催化剂为氯化亚铜 0.039g(0.4mmol),助催剂为HBr 0.014mL(0.25mmol),硫氰酸盐 为硫氰酸铵0.152g(2mmol),溶剂为乙醇5mL,压力为0.5MPa, 反应温度为80℃,反应时间为20h,其他操作同实施例1,得到α- 硫氰化环戊酮0.078g,收率55%。
1H NMR(300MHz,CDCl3):d=3.65-3.74(m,1H),2.62-2.71(m, 1H),1.95-2.45(m,5H).
III-1结构式: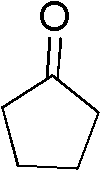
实施例4
芳香酮为邻甲基苯乙酮(I-2)0.134g(1mmol),催化剂为氯化 铜0.040g(0.3mmol),助催剂Br20.024g(0.15mmol),硫氰酸盐为 硫氰酸钠0.122g(1.5mmol),溶剂二甲亚砜5mL,压力为0.2MPa, 反应温度为60℃,反应时间为10h,其他操作同实施例1,获得α-硫 氰化邻甲基苯乙酮0.136g,收率71%。
1H NMR(300MHz,CDCl3):d=4.17-4.33(m,1H),1.24-2.81(m, 7H),1.12(d,J=1.5Hz,3H).
I-2结构式: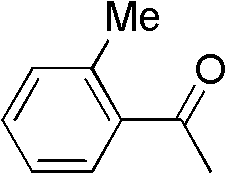
实施例5
芳香酮为2-苯基色满-4-酮(II-2)0.224g(1mmol),催化剂溴化 铜0.067g(0.3mmol),助催剂HBr为0.014mL(0.25mmol),硫氰 酸钾0.194g(1.5mmol),溶剂丙酸5mL,压力为0.4MPa,反应温 度为70℃,反应时间为12h,其他操作同实施例1,获得3-硫氰基-2- 苯基-3,4-二氢色满-4-酮0.203g,收率72%。
1H NMR(300MHz,CDCl3):d=7.96-8.06(m,1H),7.44-7.62(m, 6H),7.03-7.19(m,2H),5.49(d,J=11.3Hz,1H),4.40(d,J =11.3Hz,1H).
II-2结构式: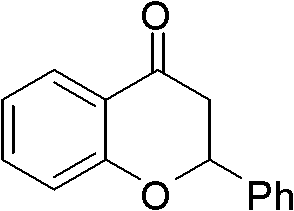
实施例6
脂肪酮为环己酮(III-2)0.098g(1mmol),催化剂醋酸铜为0.073 g(0.4mmol),助催剂Br2为0.04g(0.25mmol),硫氰酸钾0.194g(1.5 mmol),压力为0.4MPa,反应温度为80℃,反应时间为20h,其他 操作同实施例1,获得α-硫氰化环己酮0.078g,,收率58%。
1H NMR(300MHz,CDCl3):d=4.22-4.29(m,1H),2.77-2.85(m,1 H),2.61-2.68(m,1H),2.36-2.46(m,1H),1.70-2.22(m,5H).
III-2结构式: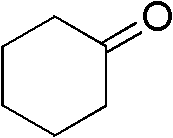
实施例7
芳香酮为邻甲氧基苯乙酮(I-3)0.150g(1mmol),催化剂为碘 化亚铜0.057g(0.3mmol),助催剂Br2为0.032g(0.2mmol),硫氰 酸盐为硫氰酸钾0.155g(1.6mmol),乙酸5mL,压力为0.2MPa,80 ℃下反应12h,其他操作同实施例1,获得α-硫氰化邻甲氧基苯乙酮 0.128g,收率62%。
1H NMR(300MHz,CDCl3):d=7.90-7.96(m,1H),7.51-7.60 (m,1H),6.97-7.10(m,2H),4.80(s,2H),4.05(s,3H).
I-3结构式: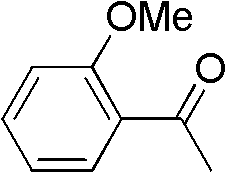
实施例8
脂肪酮为环庚酮(III-3)0.112g(1mmol),催化剂三氟醋酸铜为0.115 g(0.4mmol),助催剂Br2为0.04g(0.25mmol),硫氰酸盐为硫氰酸铵 0.152g(2mmol),溶剂丙酸5mL,压力为0.4MPa,反应温度为80℃, 反应时间为24h,其他操作同实施例1,获得α-硫氰化环己酮0.083g, 收率49%。
1H NMR(300MHz,CDCl3):d=4.42(m,1H),2.41-2.74(m,3 H),1.62-2.17(m,7H).
III-3结构式: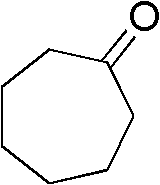
由上述的实例表明,采用本发明所提供的常压下利用氧气或空气作 氧化剂,铜盐作催化剂,氢溴酸或溴单质作助催化剂,合成α-硫氰化酮 的方法,有较宽的底物适用范围,该方法具有反应条件温和、操作易于控 制、后处理简单易行、成本低廉、反应安全、整个过程对环境友好、无污 染等特点。

 手机二维码
手机二维码 京公网安备11011302007134
京公网安备11011302007134